Warning! Note that elemental potentials taken from alloy descriptions may not work well for the pure species. This is particularly true if the elements were fit for compounds instead of being optimized separately. As with all interatomic potentials, please check to make sure that the performance is adequate for your problem.
Citation: H. Sharifi, and C.D. Wick (2025), "Developing interatomic potentials for complex concentrated alloys of Cu, Ti, Ni, Cr, Co, Al, Fe, and Mn", Computational Materials Science, 248, 113595. DOI: 10.1016/j.commatsci.2024.113595.
Abstract: Complex concentrated alloys (CCAs) are a new generation of metallic alloys composed of three or more principal elements with physical and mechanical properties that can be tuned by adjusting their compositions. The extensive compositional workspace of CCAs makes it impractical to perform a comprehensive search for a specific material property using experimental measurements. The use of computational methods can rapidly narrow down the search span, improving the efficiency of the design process. We carried out a high-throughput parameterization of modified embedded atom method (MEAM) interatomic potentials for combinations of Cu, Ti, Ni, Cr, Co, Al, Fe, and Mn using a genetic algorithm. Unary systems were parameterized based on DFT calculations and experimental results. MEAM potentials for 28 binary and 56 ternary combinations of the elements were parameterized to DFT results that were carried out with semi-automated frameworks. Specific attention was made to reproduce properties that impact compositional segregation, material strength, and mechanics.
Notes: This is a binary listing for the 2025--Sharifi-H-Wick-C-D--Fe-Mn-Ni-Ti-Cu-Cr-Co-Al potential. This potential focuses on the structural analysis of alloys including shear strength and elastic constants, dislocation dynamics and their impact on alloy strength, and the analysis of defect effects, such as voids, on material properties. However, the potential was not optimized for temperature-dependent properties and was not fit to density, thermal expansion coefficients, or thermal conductivity data.
Citation: Y. Sun, M.I. Mendelev, F. Zhang, X. Liu, B. Da, C.-Z. Wang, R.M. Wentzcovitch, and K.-M. Ho (2024), "Unveiling the effect of Ni on the formation and structure of Earth’s inner core", Proceedings of the National Academy of Sciences, 121(4), e2316477121. DOI: 10.1073/pnas.2316477121.
Abstract: Ni is the second most abundant element in the Earth’s core. Yet, its effects on the inner core’s structure and formation process are usually disregarded because of its electronic and size similarity with Fe. Using ab initio molecular dynamics simulations, we find that the bcc phase can spontaneously crystallize in liquid Ni at temperatures above Fe’s melting point at inner core pressures. The melting temperature of Ni is shown to be 700 to 800 K higher than that of Fe at 323 to 360 GPa. hcp, bcc, and liquid phase relations differ for Fe and Ni. Ni can be a bcc stabilizer for Fe at high temperatures and inner core pressures. A small amount of Ni can accelerate Fe’s crystallization at core pressures. These results suggest that Ni may substantially impact the structure and formation process of the solid inner core.
Notes: The potential was employed in the TI calculations in the above reference. It can be used as an initial approximation for MD simulations under the Earth’s inner core conditions.
Citation: C. Wu, B.-J. Lee, and X. Su (2017), "Modified embedded-atom interatomic potential for Fe-Ni, Cr-Ni and Fe-Cr-Ni systems", Calphad, 57, 98-106. DOI: 10.1016/j.calphad.2017.03.007.
Abstract: A semi-empirical interatomic potential formalism, the second-nearest-neighbor modified embedded-atom method (2NN MEAM), has been applied to obtaining interatomic potentials for the Fe-Ni, Cr-Ni and Fe-Cr-Ni systems using previously developed MEAM potentials of Fe and Ni and a newly revised potential of Cr. The potential parameters were determined by fitting the experimental data on the enthalpy of formation or mixing, lattice parameter and elastic constant. The present potentials generally reproduced the fundamental physical properties of the Fe-Ni and Cr-Ni alloys. The enthalpy of formation or mixing of the disordered phase at finite temperature and the enthalpy of mixing of the liquid phase are reasonable in agreements with experiment data and CALPHAD calculations. The potentials can be combined with already-developed MEAM potentials to describe Fe-Cr-Ni-based multicomponent alloys. Moreover, the average diffusivities in the unary, some binary and ternary alloys were simulated based on present potential. Good agreement is obtained in comparison with experimental data.
Citation: G. Bonny, R.C. Pasianot, and L. Malerba (2009), "Fe-Ni many-body potential for metallurgical applications", Modelling and Simulation in Materials Science and Engineering, 17(2), 025010. DOI: 10.1088/0965-0393/17/2/025010.
Abstract: A many-body interatomic potential for the Fe–Ni system is fitted, capable of describing both the ferritic and austenitic phase. The Fe–Ni system exhibits two stable ordered intermetallic phases, namely, L10 FeNi and L12 FeNi3, that are key issues to be tackled when creating a Fe–Ni potential consistent with thermodynamics. A procedure, based on a rigid lattice Ising model and the theory of correlation functions space, is developed to address all the intermetallics that are possible ground states of the system. While controlling the ground states of the system, the mixing enthalpy and defect properties were fitted. Both bcc and fcc defect properties are compared with density functional theory calculations and other potentials found in the literature. Finally, the potential is thermodynamically validated by constructing the alloy phase diagram. It is shown that the experimental phase diagram is reproduced reasonably well and that our potential gives a globally improved description of the Fe–Ni system in the whole concentration range with respect to the potentials found in the literature.
See Computed Properties Notes: Listing found at https://openkim.org. This KIM potential is based on the files from 2009--Bonny-G--Fe-Ni--LAMMPS--ipr1. Link(s):
Citation: Y. Mishin, M.J. Mehl, and D.A. Papaconstantopoulos (2005), "Phase stability in the Fe-Ni system: Investigation by first-principles calculations and atomistic simulations", Acta Materialia, 53(15), 4029-4041. DOI: 10.1016/j.actamat.2005.05.001.
Abstract: First-principles calculations of the energy of various crystal structures of Fe, Ni and ordered Fe–Ni compounds with different stoichiometries have been performed by the linearized augmented plane wave (LAPW) method in the generalized gradient approximation. The most stable compounds are L12–Ni3Fe, L10–FeNi, C11f–Ni2Fe and C11f–Fe2Ni. The L12-Ni3Fe compound has the largest negative formation energy, which is consistent with the experimental Fe–Ni phase diagram. The L10–FeNi compound has also been observed experimentally in meteorite samples as a metastable phase. It is suggested here that the C11f compounds could also form in Fe–Ni alloys at low temperatures. A new semi-empirical interatomic potential has been developed for the Fe–Ni system by fitting to experimental data and the results of the LAPW calculations. Recognizing the significance of the covalent component of bonding in this system, the potential is based on the embedded-atom method (EAM) but additionally includes a bond-angle dependence. In comparison with the existing modified EAM method, our potential form is simpler, extends interactions to several (3–5) coordination shells and replaces the screening procedure by a smooth cutoff of the potential functions. The potential reproduces a variety of properties of Fe and Ni with a reasonable accuracy. It also reproduces all stability trends across the Fe–Ni system established by the LAPW calculations. The potential can be useful in atomistic simulations of the phases of the Fe–Ni system.
Notes: These files were provided by Yuri Mishin (George Mason University) and posted on 22 Dec. 2009. Prof. Mishin requested the following note be included: "The equation appearing in the Appendix on page 4040 contains a typing error: the sign before 1/3 in the last line must be negative." He provided the corrected equation for the angular-dependent force contributions in ADP_Forces.jpg or ADP_Forces.pdf. File(s):
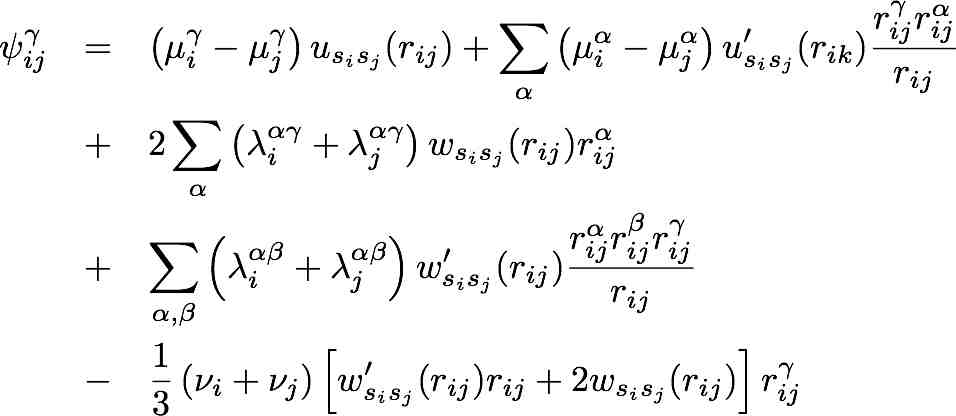{kind=link}