-
Citation: D. Schopf, P. Brommer, B. Frigan, and H.-R. Trebin (2012), "Embedded atom method potentials for Al-Pd-Mn phases", Physical Review B 85(5), 054201. DOI: 10.1103/physrevb.85.054201.Abstract: A novel embedded atom method (EAM) potential for the Ξ phases of Al-Pd-Mn has been determined with the force-matching method. Different combinations of analytic functions were tested for the pair and transfer part. The best results are obtained if one allows for oscillations on two different length scales. These potentials stabilize structure models of the Ξ phases and describe their energy with high accuracy. Simulations at temperatures up to 1200 K show very good agreement with ab initio results with respect to stability and dynamics of the system.
Related Models: -
LAMMPS pair_style eam/alloy (2012--Schopf-D--Al-Mn-Pd--LAMMPS--ipr1)See Computed Properties
Notes: This version is compatible with LAMMPS. UPDATE 11 June 2012: The version posted on 26 April 2012 had an extra line in the header and did not work with LAMMPS. This was brought to our attention by Daniel Schopf and the correct version has been posted. Original note: This file was provided by Daniel Schopf (Stuttgart University) and posted with his permission on 26 April 2012.
File(s): -
IMD option EAM (2012--Schopf-D--Al-Mn-Pd--IMD--ipr1)Notes: These files were provided by Daniel Schopf.
File(s):F(ρ): AlMnPd_Schopf_2012_imd_F.imd.pt
φ(r): AlMnPd_Schopf_2012_imd_phi.imd.pt
ρ(r): AlMnPd_Schopf_2012_imd_rho.imd.pt -
OpenKIM (MO_137572817842)See Computed Properties
Notes: Listing found at https://openkim.org. This KIM potential is based on the files from 2012--Schopf-D--Al-Mn-Pd--LAMMPS--ipr1.
Link(s): -
OpenKIM (MO_878712978062)See Computed Properties
Notes: Listing found at https://openkim.org. This KIM potential is based on the same files as 2012--Schopf-D--Al-Mn-Pd--IMD--ipr1.
Link(s):
Implementation Information
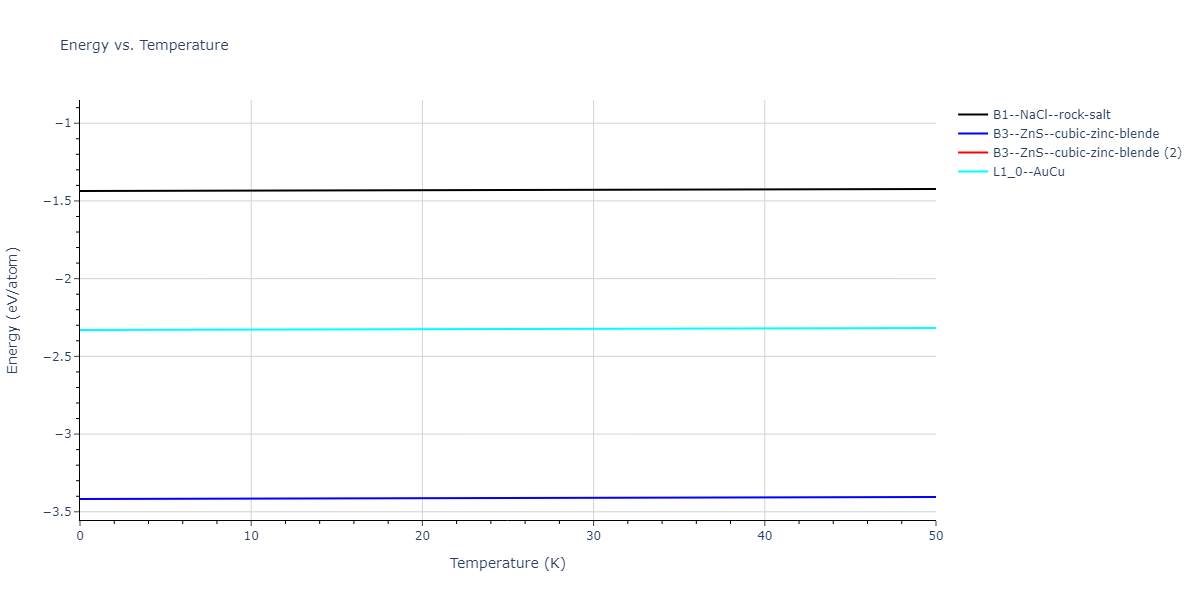
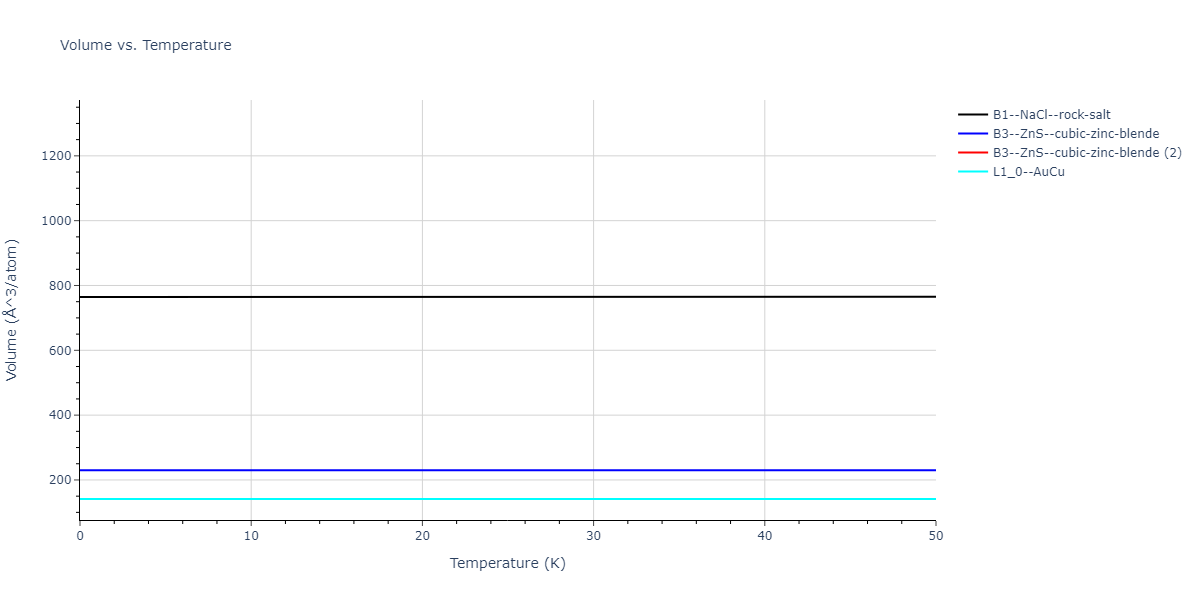
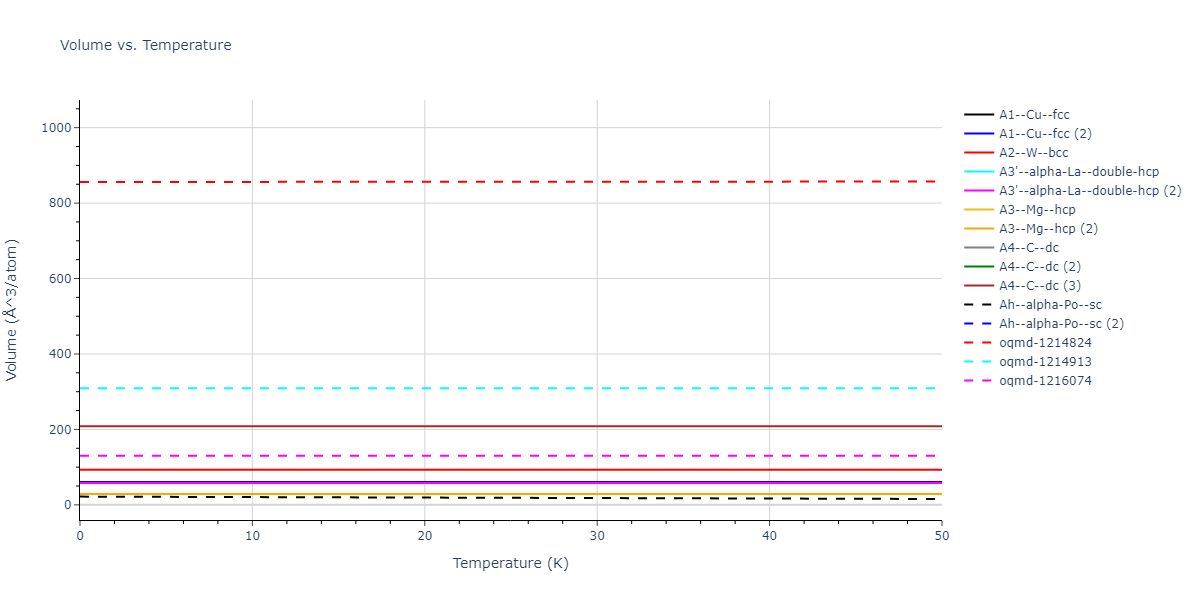
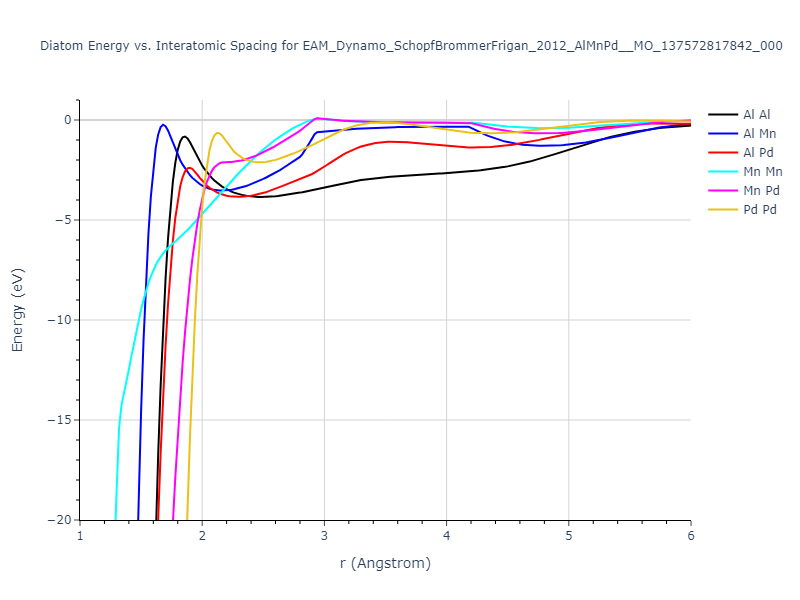
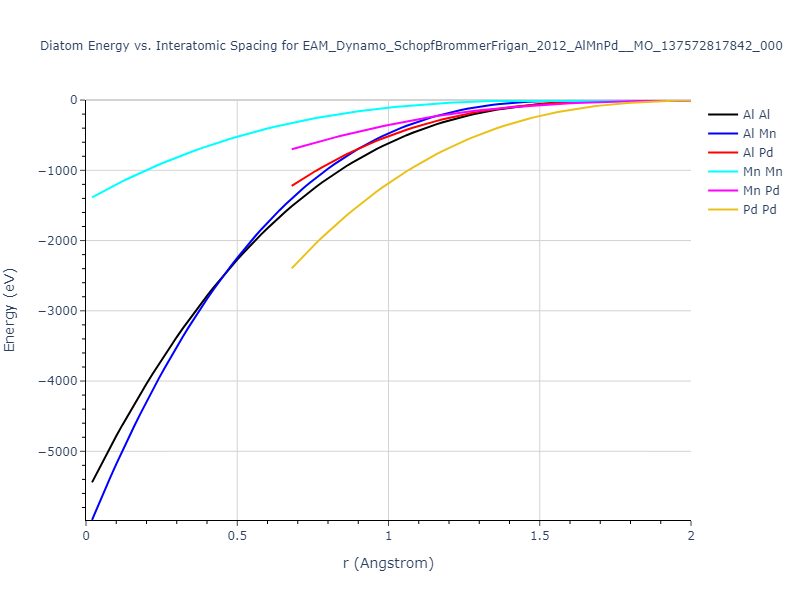
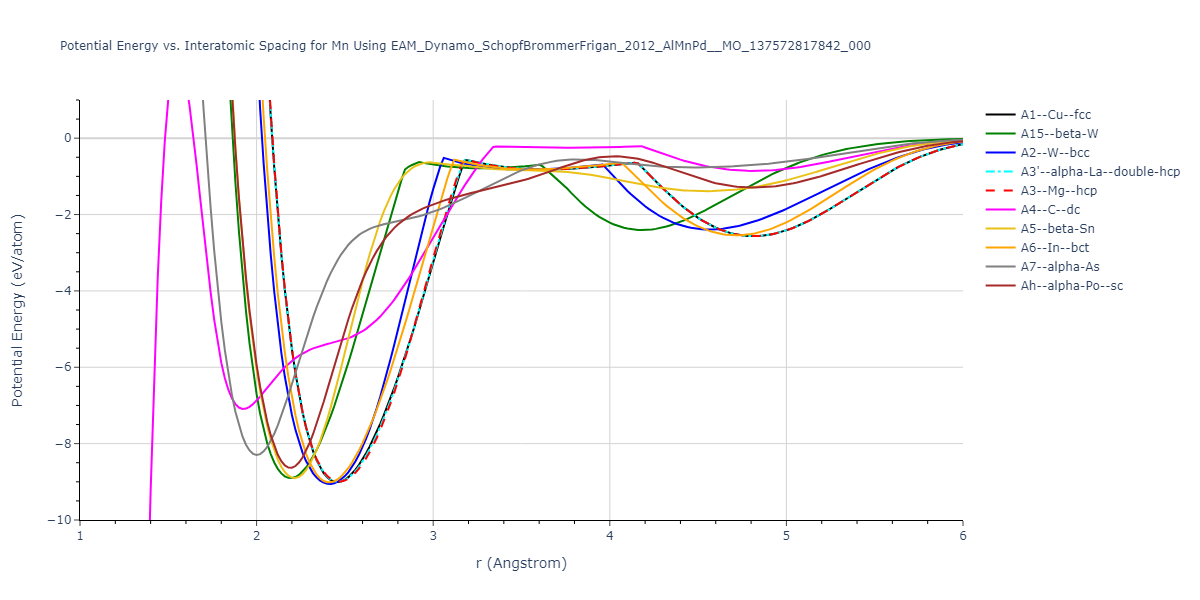
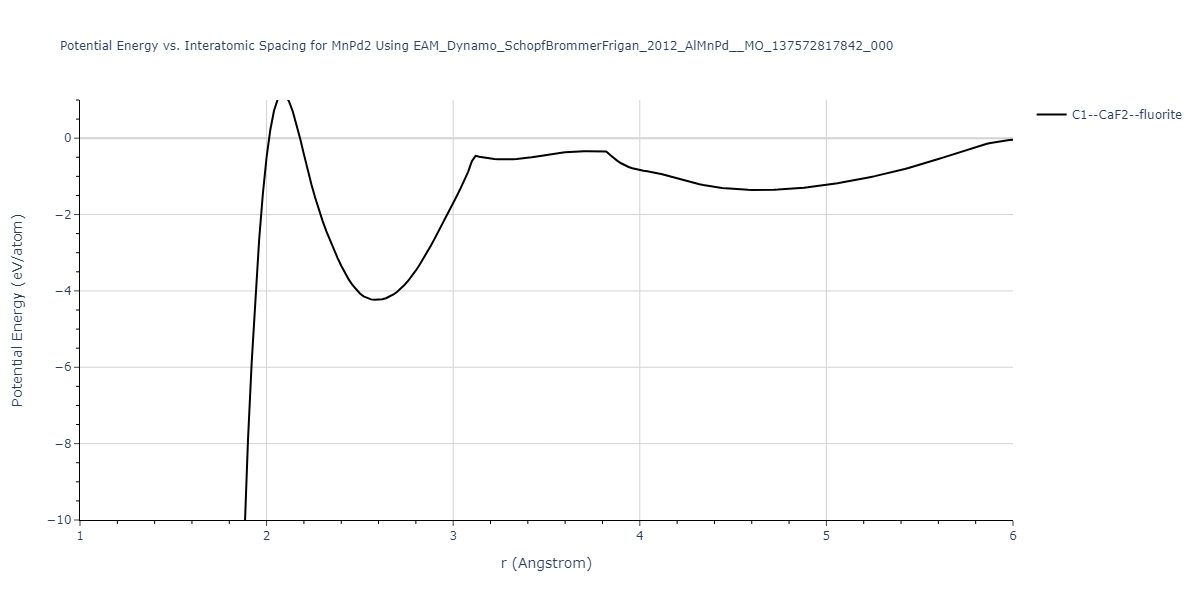
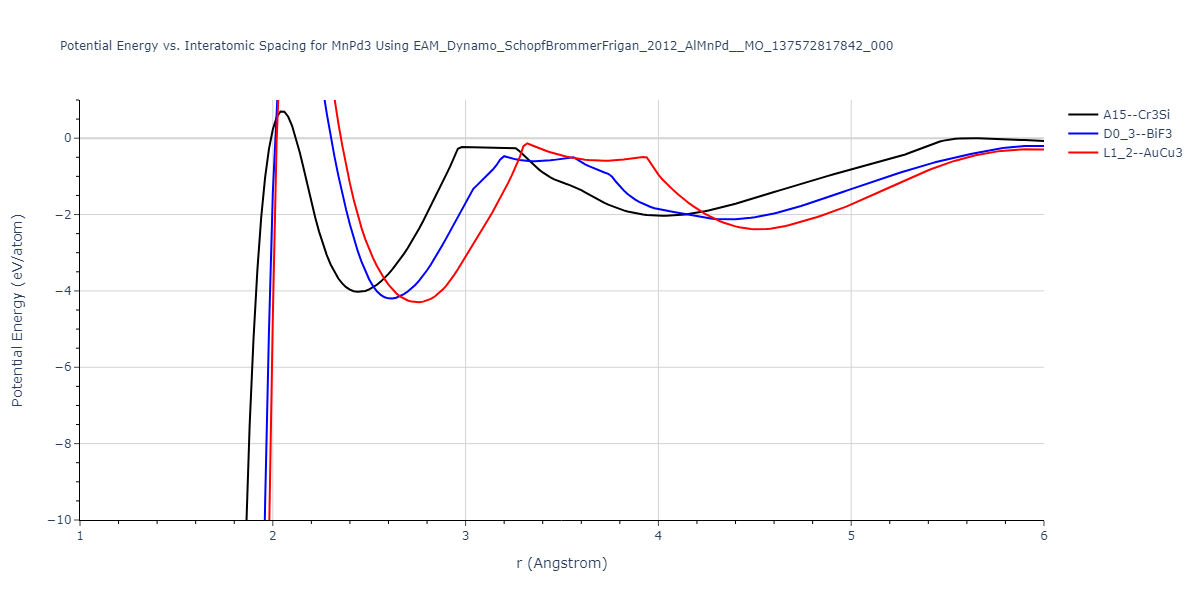
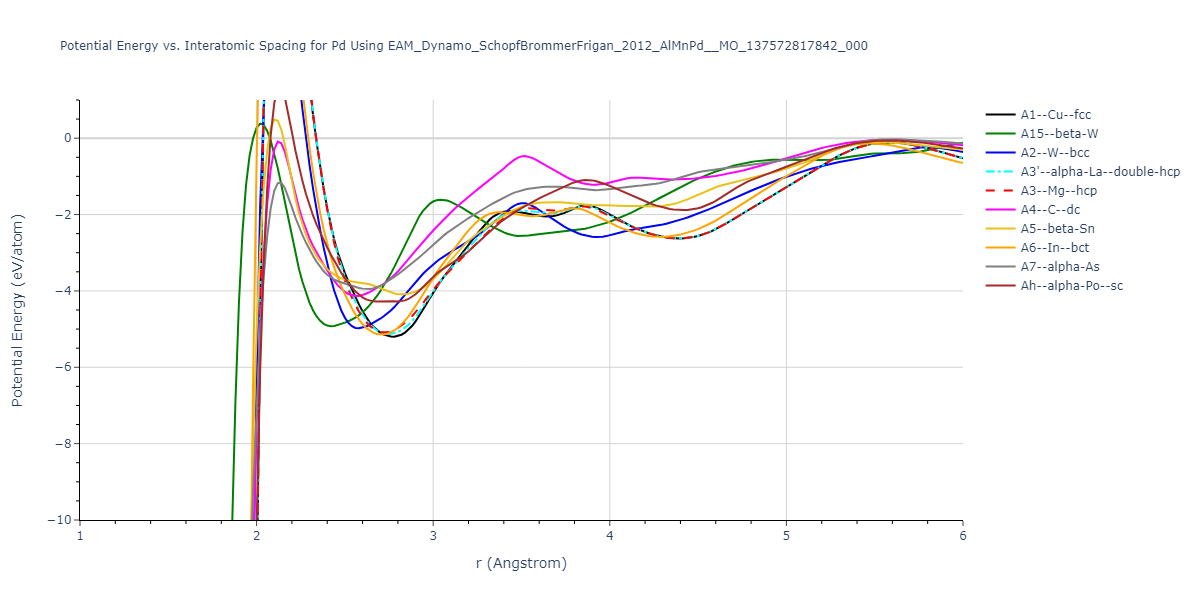
Diatom Energy vs. Interatomic Spacing
Plots of the potential energy vs interatomic spacing, r, are shown below for all diatom sets associated with the interatomic potential. This calculation provides insights into the functional form of the potential's two-body interactions. A system consisting of only two atoms is created, and the potential energy is evaluated for the atoms separated by 0.02 Å <= r <= 6.0> Å in intervals of 0.02 Å. Two plots are shown: one for the "standard" interaction distance range, and one for small values of r. The small r plot is useful for determining whether the potential is suitable for radiation studies.
The calculation method used is available as the iprPy diatom_scan calculation method.
Clicking on the image of a plot will open an interactive version of it in a new tab. The underlying data for the plots can be downloaded by clicking on the links above each plot.
Notes and Disclaimers:
- These values are meant to be guidelines for comparing potentials, not the absolute values for any potential's properties. Values listed here may change if the calculation methods are updated due to improvements/corrections. Variations in the values may occur for variations in calculation methods, simulation software and implementations of the interatomic potentials.
- As this calculation only involves two atoms, it neglects any multi-body interactions that may be important in molecules, liquids and crystals.
- NIST disclaimer
Version Information:
- 2019-11-14. Maximum value range on the shortrange plots are now limited to "expected" levels as details are otherwise lost.
- 2019-08-07. Plots added.
Click on plot to load interactive version
Click on plot to load interactive version
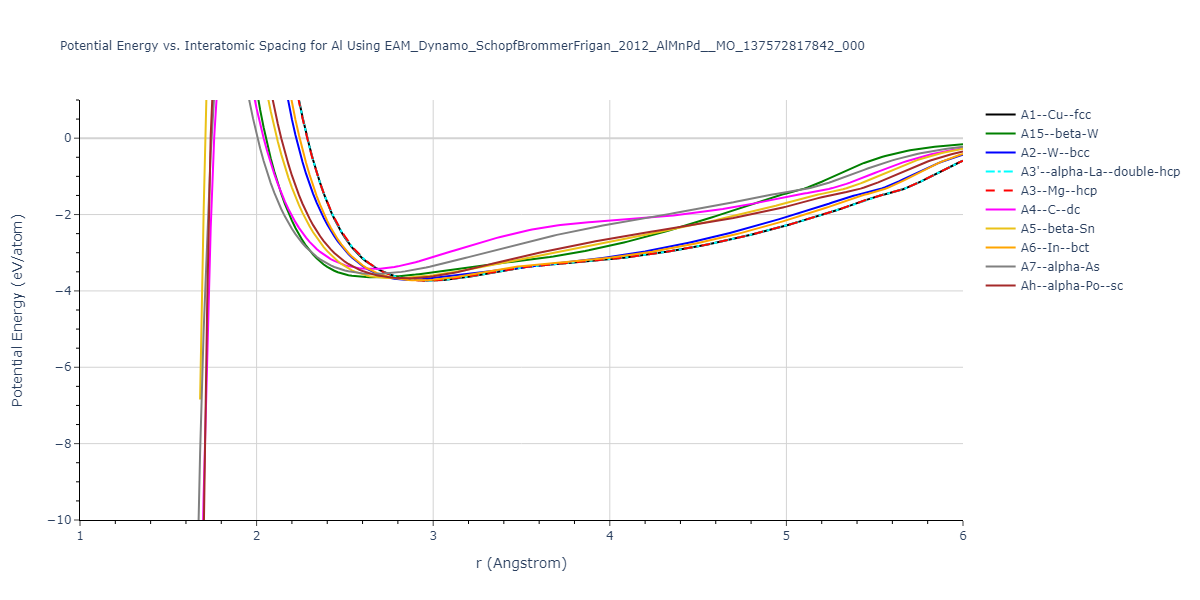
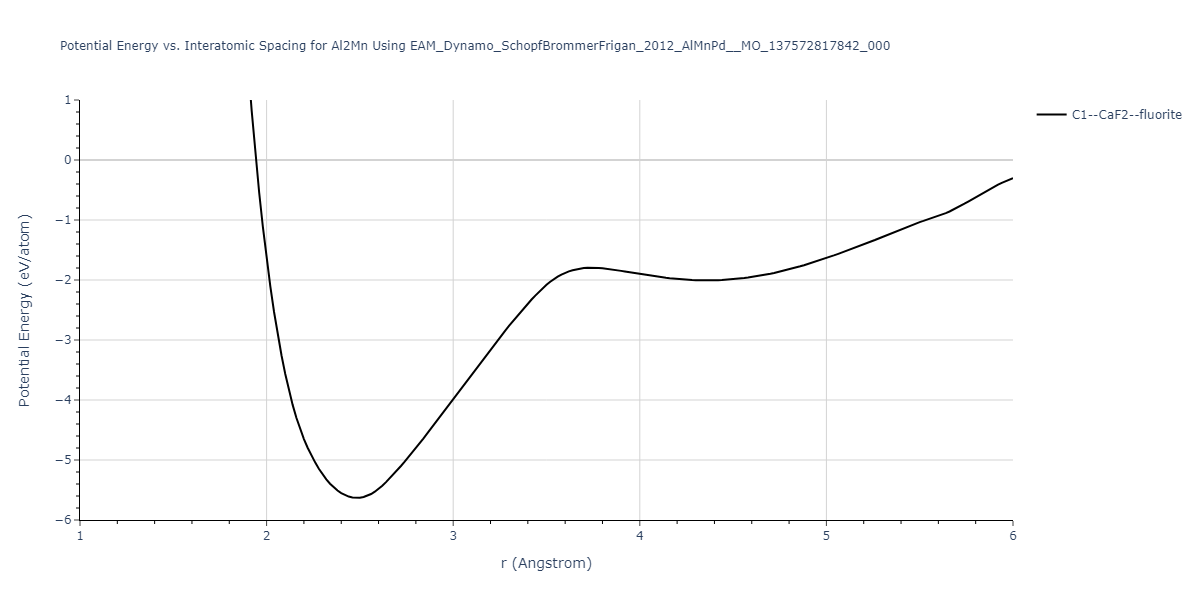
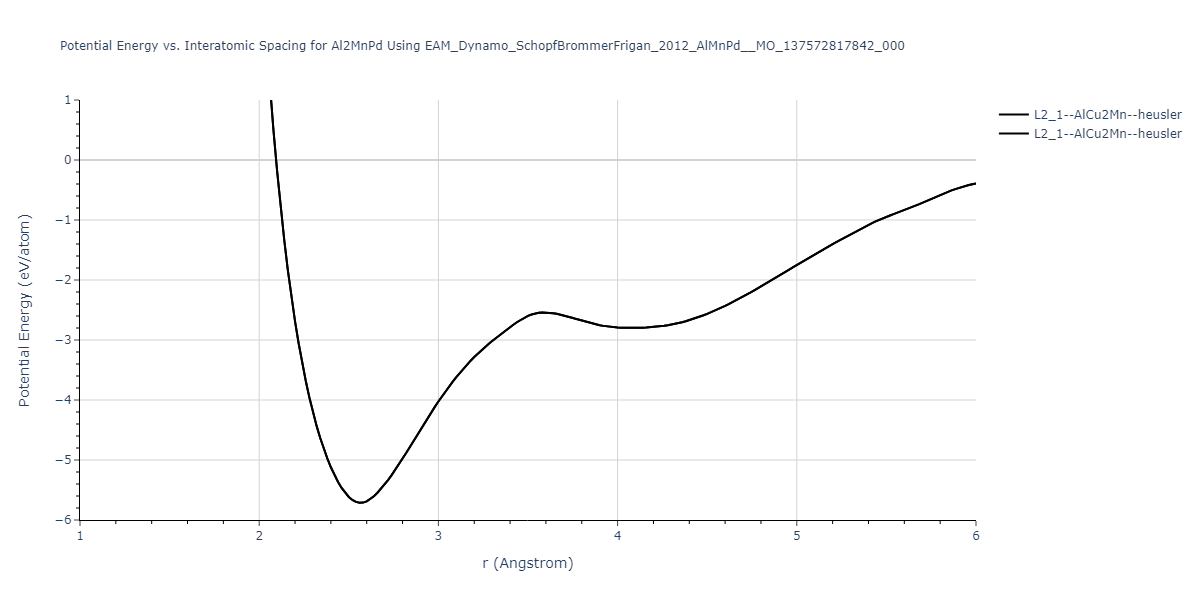
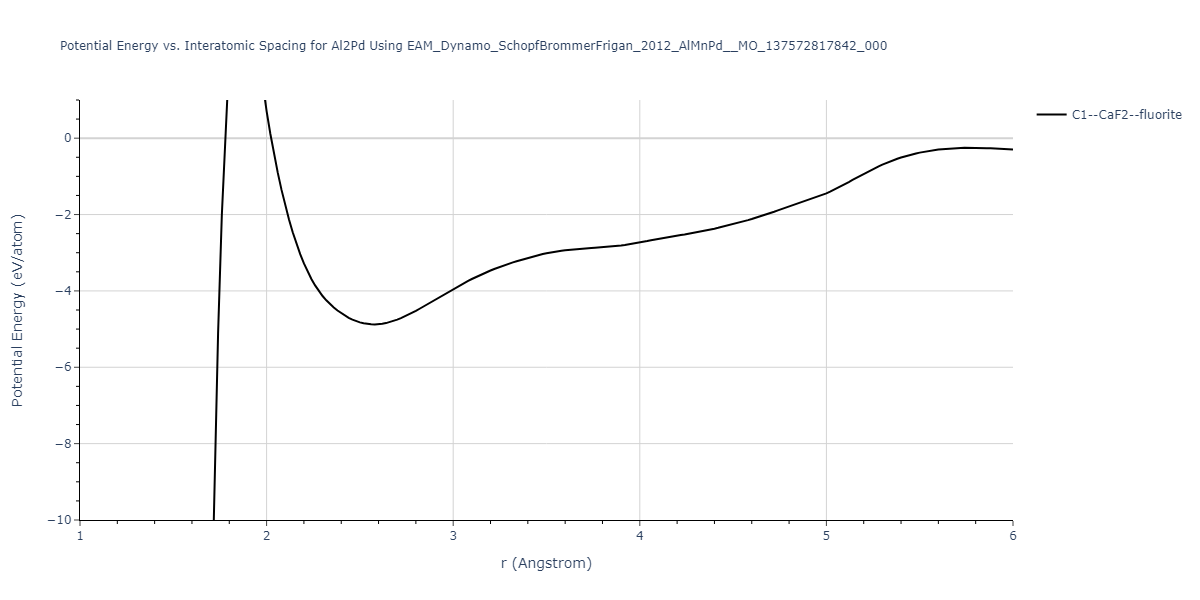
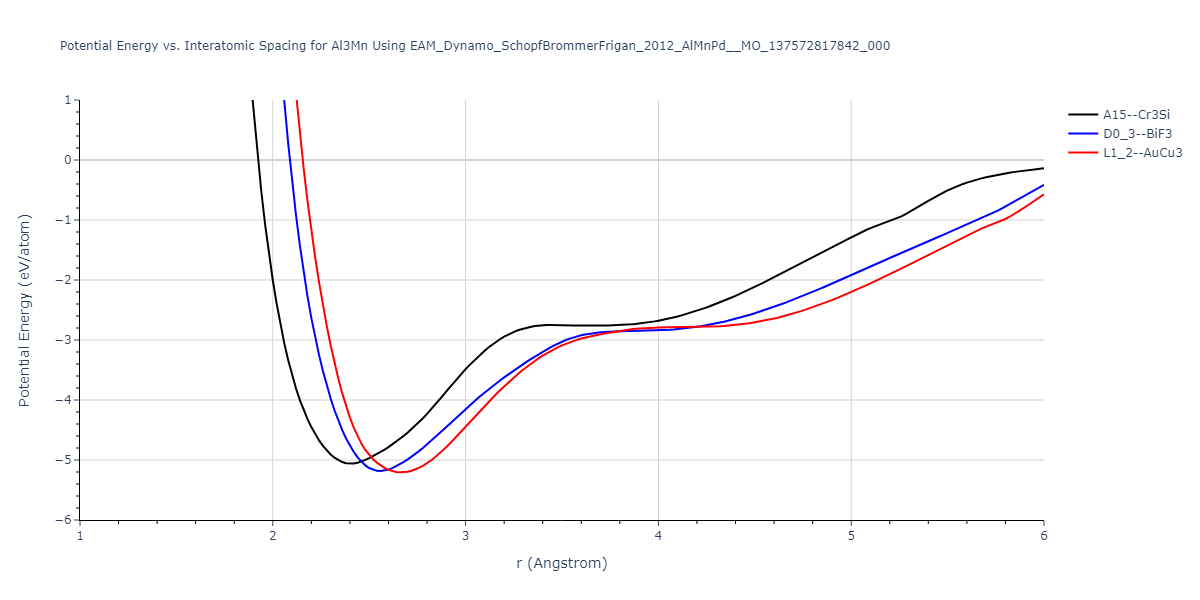
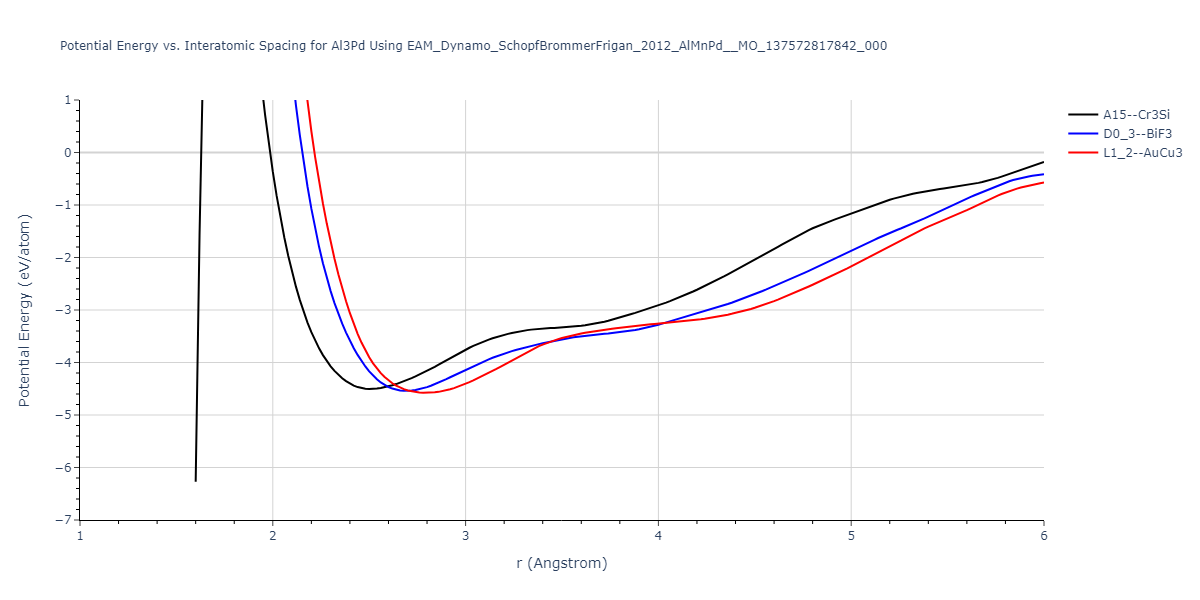
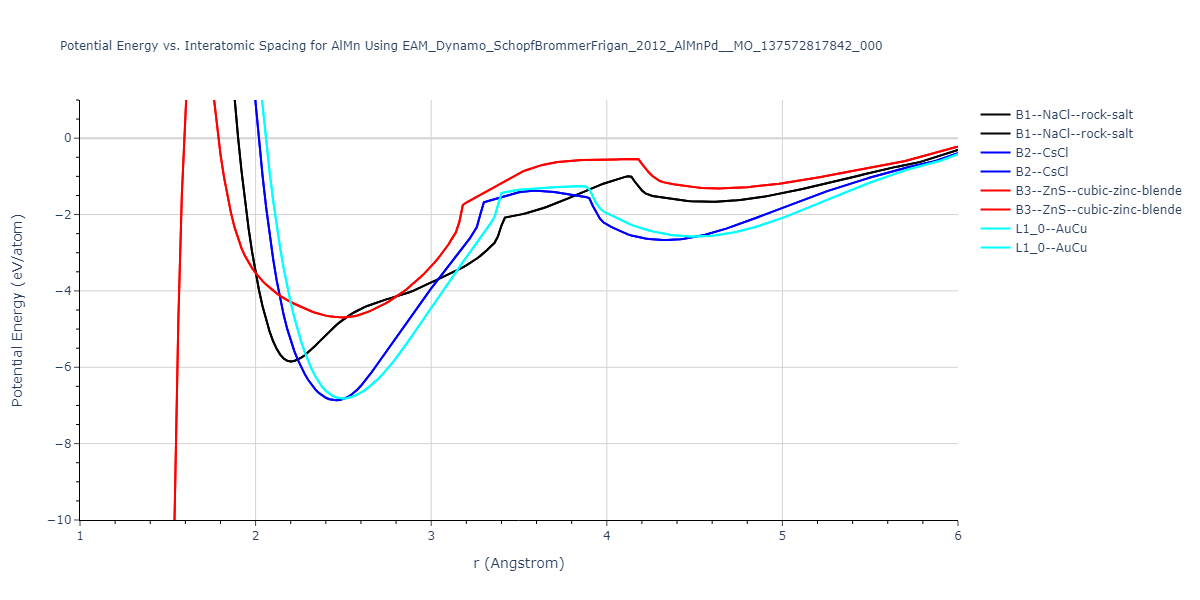
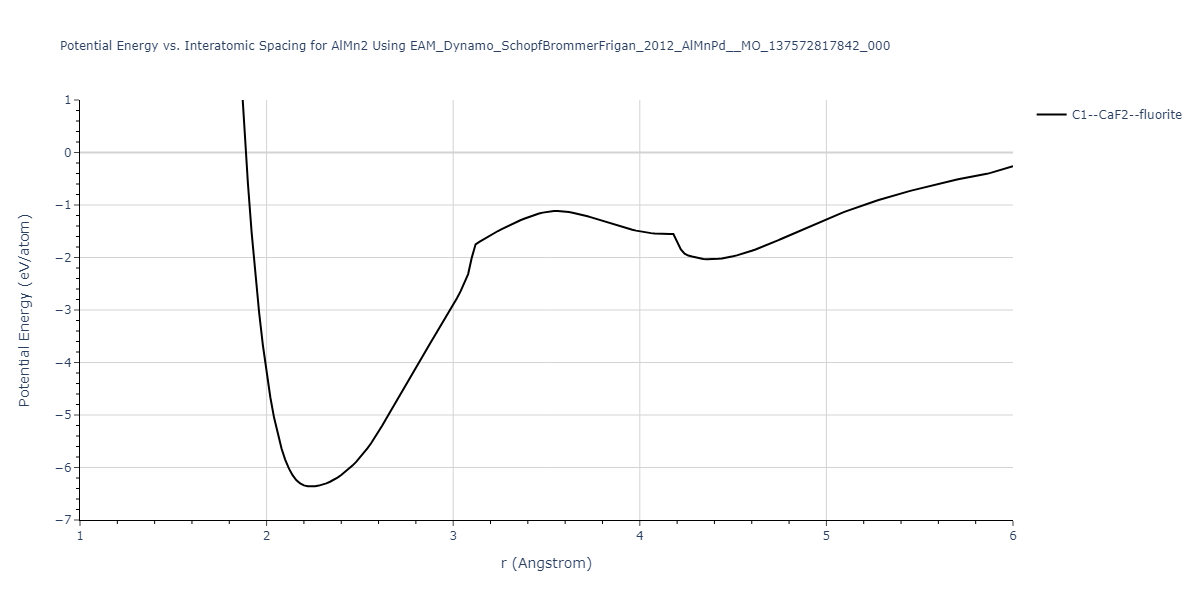
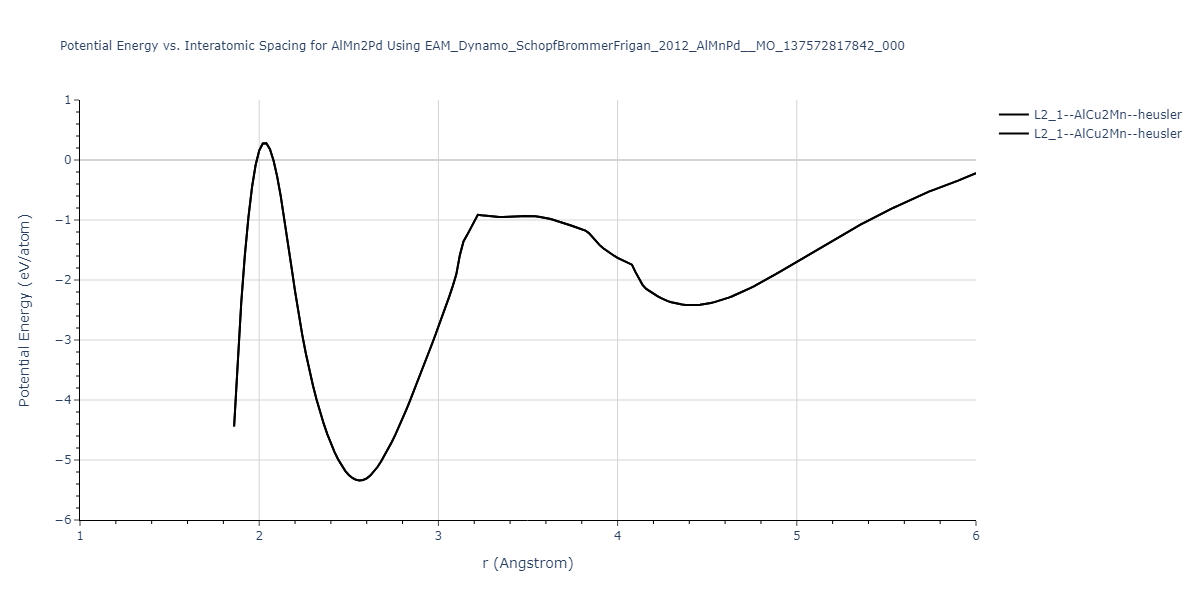
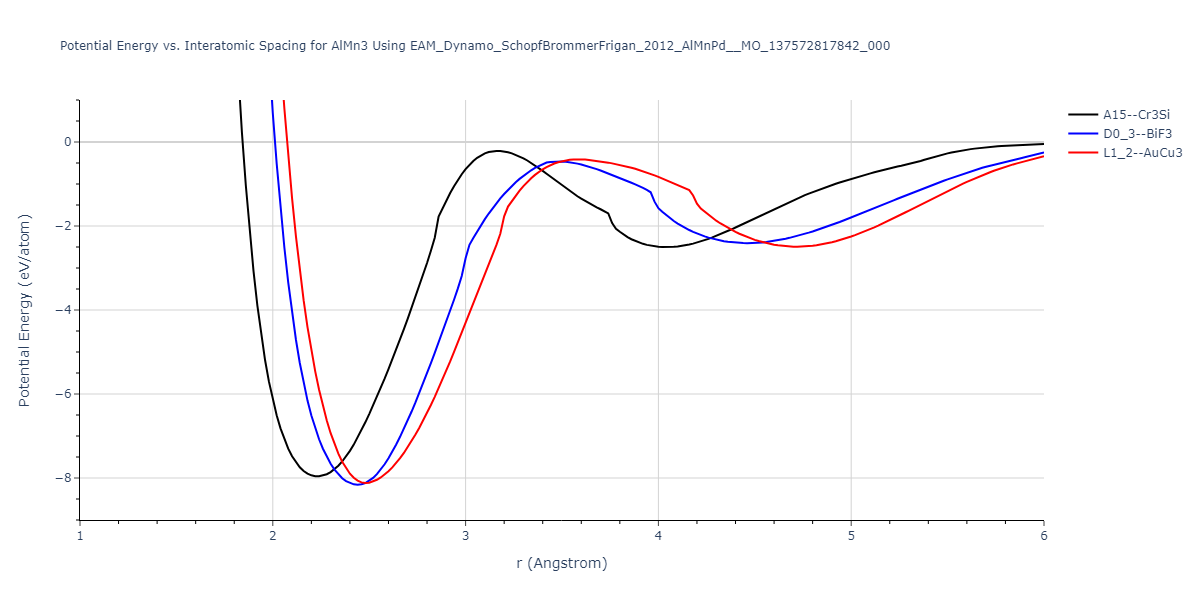
Cohesive Energy vs. Interatomic Spacing
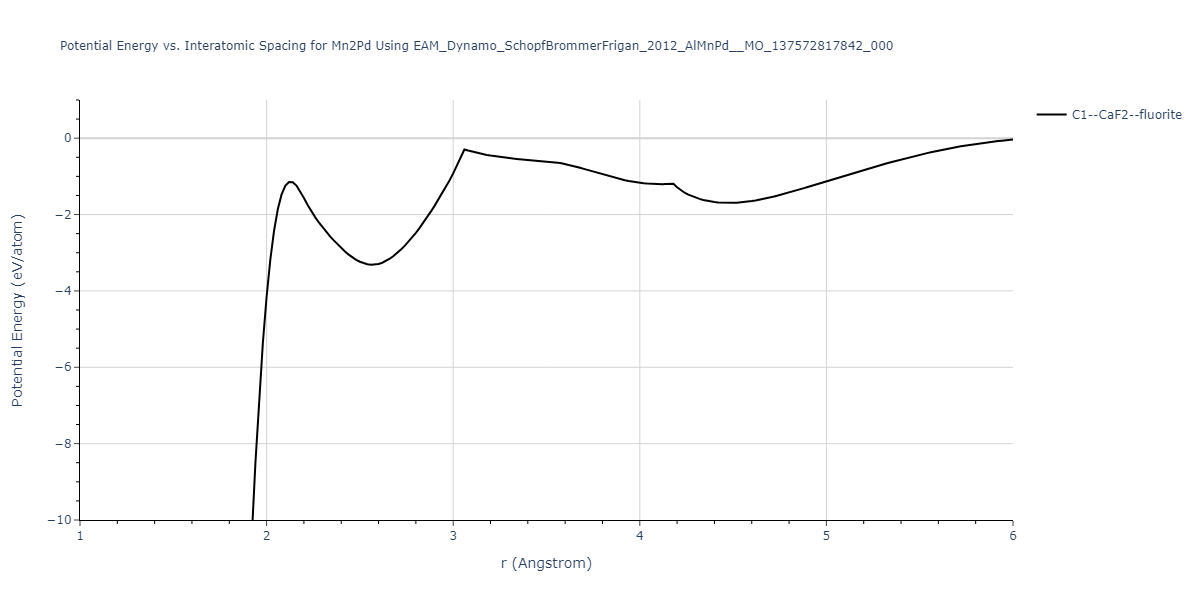
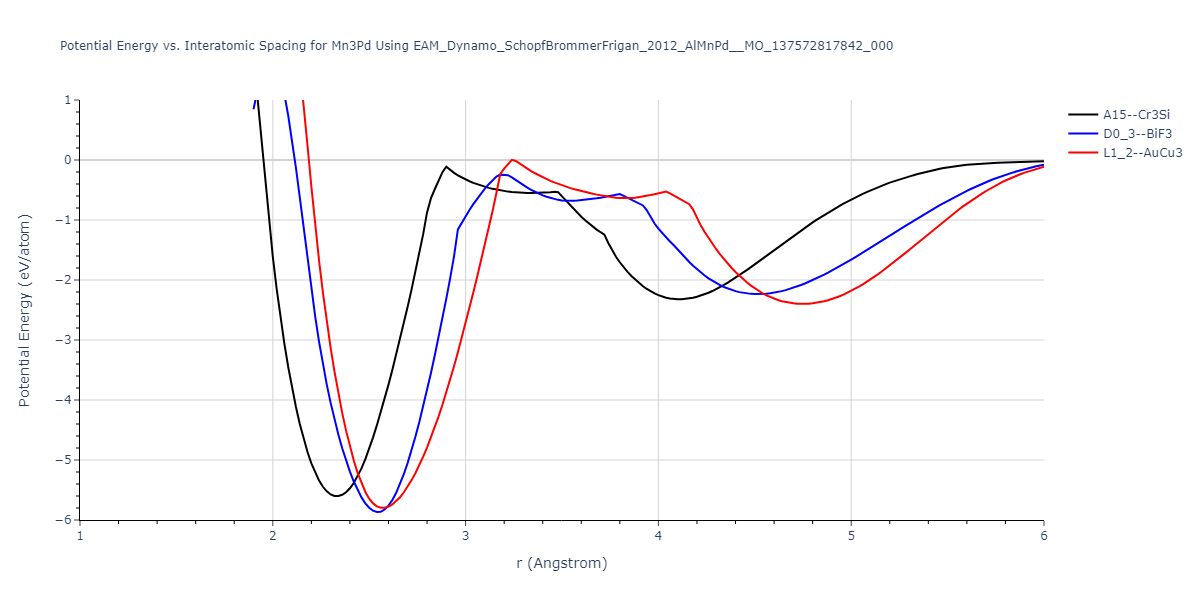
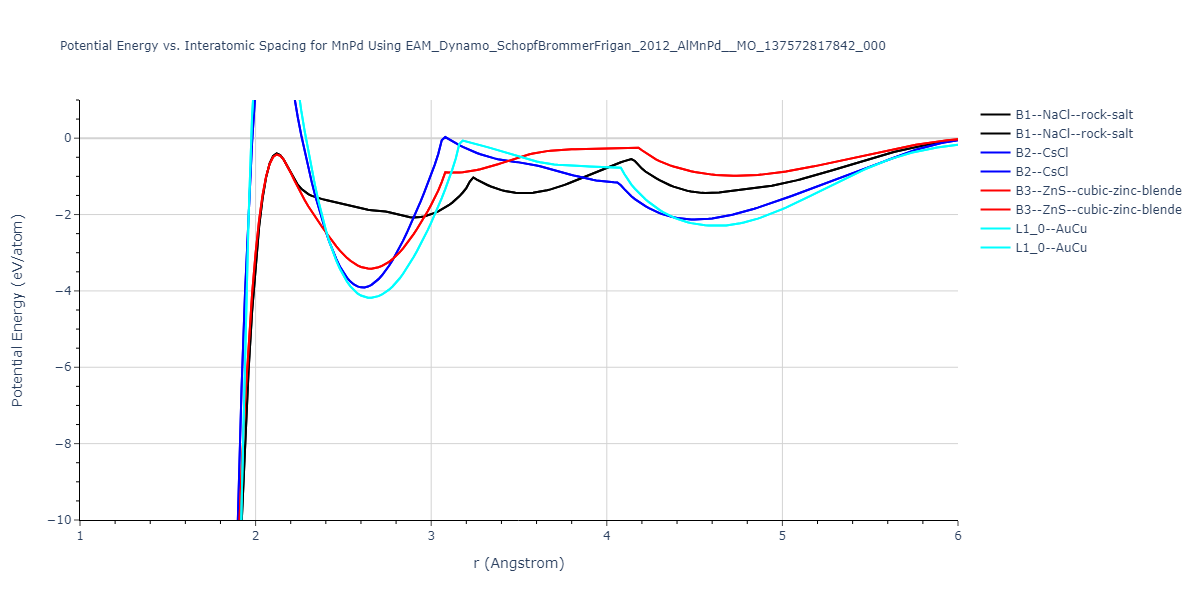
Plots of potential energy vs interatomic spacing, r, are shown below for a number of crystal structures. The structures are generated based on the ideal atomic positions and b/a and c/a lattice parameter ratios for a given crystal prototype. The size of the system is then uniformly scaled, and the energy calculated without relaxing the system. To obtain these plots, values of r are evaluated every 0.02 Å up to 6 Å.
The calculation method used is available as the iprPy E_vs_r_scan calculation method.
Clicking on the image of a plot will open an interactive version of it in a new tab. The underlying data for the plots can be downloaded by clicking on the links above each plot.
Notes and Disclaimers:
- These values are meant to be guidelines for comparing potentials, not the absolute values for any potential's properties. Values listed here may change if the calculation methods are updated due to improvements/corrections. Variations in the values may occur for variations in calculation methods, simulation software and implementations of the interatomic potentials.
- The minima identified by this calculation do not guarantee that the associated crystal structures will be stable since no relaxation is performed.
- NIST disclaimer
Version Information:
- 2020-12-18. Descriptions, tables and plots updated to reflect that the energy values are the measuredper atom potential energy rather than cohesive energy as some potentials have non-zero isolated atom energies.
- 2019-02-04. Values regenerated with even r spacings of 0.02 Å, and now include values less than 2 Å when possible. Updated calculation method and parameters enhance compatibility with more potential styles.
- 2019-04-26. Results for hcp, double hcp, α-As and L10 prototypes regenerated from different unit cell representations. Only α-As results show noticable (>1e-5 eV) difference due to using a different coordinate for Wykoff site c position.
- 2018-06-13. Values for MEAM potentials corrected. Dynamic versions of the plots moved to separate pages to improve page loading. Cosmetic changes to how data is shown and updates to the documentation.
- 2017-01-11. Replaced png pictures with interactive Bokeh plots. Data regenerated with 200 values of r instead of 300.
- 2016-09-28. Plots for binary structures added. Data and plots for elemental structures regenerated. Data values match the values of the previous version. Data table formatting slightly changed to increase precision and ensure spaces between large values. Composition added to plot title and structure names made longer.
- 2016-04-07. Plots for elemental structures added.
Crystal Structure Predictions
Computed lattice constants and cohesive/potential energies are displayed for a variety of crystal structures. The values displayed here are obtained using the following process.
- Initial crystal structure guesses are taken from:
- The iprPy E_vs_r_scan calculation results (shown above) by identifying all energy minima along the measured curves for a given crystal prototype + composition.
- Structures in the Materials Project and OQMD DFT databases.
- All initial guesses are relaxed using three independent methods using a 10x10x10 supercell:
- "box": The system's lattice constants are adjusted to zero pressure without internal relaxations using the iprPy relax_box calculation with a strainrange of 1e-6.
- "static": The system's lattice and atomic positions are statically relaxed using the iprPy relax_static calculation with a minimization force tolerance of 1e-10 eV/Angstrom.
- "dynamic": The system's lattice and atomic positions are dynamically relaxed for 10000 timesteps of 0.01 ps using the iprPy relax_dynamic calculation with an nph integration plus Langevin thermostat. The final configuration is then used as input in running an iprPy relax_static calculation with a minimization force tolerance of 1e-10 eV/Angstrom.
- The relaxed structures obtained from #2 are then evaluated using the spglib package to identify an ideal crystal unit cell based on the results.
- The space group information of the ideal unit cells is compared to the space group information of the corresponding reference structures to identify which structures transformed upon relaxation. The structures that did not transform to a different structure are listed in the table(s) below. The "method" field indicates the most rigorous relaxation method where the structure did not transform. The space group information is also used to match the DFT reference structures to the used prototype, where possible.
- The cohesive energy, Ecoh, is calculated from the measured potential energy per atom, Epot$, by subtracting the isolated energy averaged across all atoms in the unit cell. The isolated atom energies of each species model is obtained either by evaluating a single atom atomic configuration, or by identifying the first energy plateau from the diatom scan calculations for r > 2 Å.
The calculation methods used are implemented into iprPy as the following calculation styles
Notes and Disclaimers:
- These values are meant to be guidelines for comparing potentials, not the absolute values for any potential's properties. Values listed here may change if the calculation methods are updated due to improvements/corrections. Variations in the values may occur for variations in calculation methods, simulation software and implementations of the interatomic potentials.
- The presence of any structures in this list does not guarantee that those structures are stable. Also, the lowest energy structure may not be included in this list.
- Multiple values for the same crystal structure but different lattice constants are possible. This is because multiple energy minima are possible for a given structure and interatomic potential. Having multiple energy minima for a structure does not necessarily make the potential "bad" as unwanted configurations may be unstable or correspond to conditions that may not be relevant to the problem of interest (eg. very high strains).
- NIST disclaimer
Version Information:
- 2025-07-02. All "mp-" reference structure calculations were re-relaxed using the updated Materials Project database rather than the original database structures. Also, a bug was fixed that caused the "static" relaxations to occasionally throw unnecessary errors. This was fixed and all affected calculations were reset and performed again.
- 2022-05-27. The "box" method results have all been redone with an updated methodology more suited for non-orthogonal systems.
- 2020-12-18. Cohesive energies have been corrected by making them relative to the energies of the isolated atoms. The previous cohesive energy values are now listed as the potential energies.
- 2019-06-07. Structures with positive or near zero cohesive energies removed from the display tables. All values still present in the raw data files.
- 2019-04-26. Calculations now computed for each implementation. Results for hcp, double hcp, α-As and L10 prototypes regenerated from different unit cell representations.
- 2018-06-14. Methodology completely changed affecting how the information is displayed. Calculations involving MEAM potentials corrected.
- 2016-09-28. Values for simple compounds added. All identified energy minima for each structure are listed. The existing elemental data was regenerated. Most values are consistent with before, but some differences have been noted. Specifically, variations are seen with some values for potentials where the elastic constants don't vary smoothly near the equilibrium state. Additionally, the inclusion of some high-energy structures has changed based on new criteria for identifying when structures have relaxed to another structure.
- 2016-04-07. Values for elemental crystal structures added. Only values for the global energy minimum of each unique structure given.
Download raw data (including filtered results)
Reference structure matches:
A1--Cu--fcc = mp-134, oqmd-8100, oqmd-1214503
A15--beta-W = oqmd-1214948, oqmd-1280406
A2--W--bcc = mp-998860, oqmd-1215126
A3'--alpha-La--double-hcp = mp-1183144, oqmd-1215394
A3--Mg--hcp = oqmd-1215215, oqmd-1215304
A4--C--dc = oqmd-1215483
A5--beta-Sn = oqmd-1215572
A6--In--bct = oqmd-1215661
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
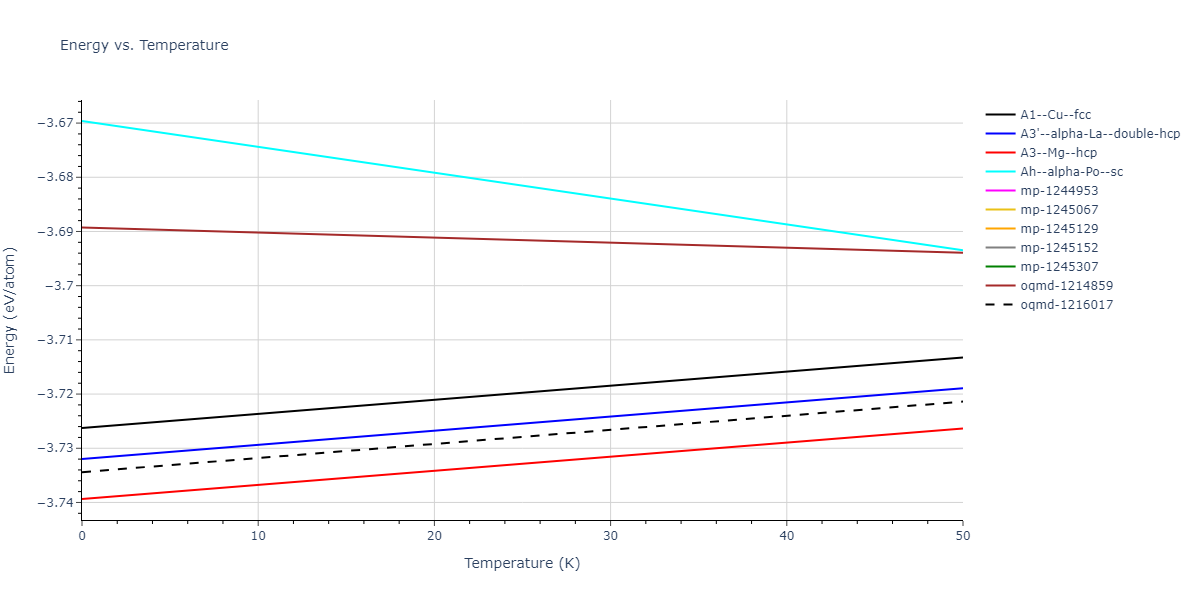
| A3--Mg--hcp | dynamic | -3.6433 | -3.7394 | 3.0299 | 3.0299 | 4.6558 | 90.0 | 90.0 | 120.0 |
| A3--Mg--hcp | box | -3.6433 | -3.7394 | 3.0298 | 3.0298 | 4.6558 | 90.0 | 90.0 | 120.0 |
| oqmd-1216017 | dynamic | -3.6384 | -3.7344 | 3.0185 | 3.0185 | 21.1285 | 90.0 | 90.0 | 120.0 |
| oqmd-1216017 | static | -3.6383 | -3.7343 | 3.0199 | 3.0199 | 21.1056 | 90.0 | 90.0 | 120.0 |
| A3'--alpha-La--double-hcp | dynamic | -3.6359 | -3.732 | 3.0124 | 3.0124 | 9.4341 | 90.0 | 90.0 | 120.0 |
| A1--Cu--fcc | dynamic | -3.6302 | -3.7263 | 4.2107 | 4.2107 | 4.2107 | 90.0 | 90.0 | 90.0 |
| oqmd-1215037 | box | -3.6116 | -3.7076 | 2.91 | 4.9554 | 10.7271 | 90.0 | 90.0 | 90.0 |
| A2--W--bcc | static | -3.611 | -3.7071 | 3.3055 | 3.3055 | 3.3055 | 90.0 | 90.0 | 90.0 |
| mp-1245129 | dynamic | -3.6099 | -3.7059 | 11.4995 | 12.3416 | 13.9675 | 93.3 | 92.5 | 100.4 |
| mp-1245307 | dynamic | -3.6073 | -3.7034 | 12.2259 | 12.2953 | 13.0213 | 84.4 | 87.6 | 89.0 |
| mp-1245152 | dynamic | -3.6065 | -3.7025 | 12.4613 | 12.5707 | 12.6225 | 90.6 | 91.4 | 97.7 |
| mp-1244953 | dynamic | -3.6053 | -3.7014 | 12.4296 | 12.5475 | 12.8161 | 85.5 | 78.8 | 85.4 |
| mp-1245067 | dynamic | -3.6043 | -3.7004 | 12.2642 | 12.7106 | 12.9761 | 89.8 | 84.2 | 77.8 |
| mp-1245129 | static | -3.6033 | -3.6993 | 11.9058 | 12.6837 | 13.1647 | 93.3 | 92.1 | 100.2 |
| mp-1245152 | static | -3.6027 | -3.6988 | 12.279 | 12.6802 | 12.6899 | 91.9 | 93.6 | 97.4 |
| mp-1244953 | static | -3.6016 | -3.6976 | 12.4797 | 12.4873 | 12.7914 | 79.1 | 86.5 | 84.9 |
| mp-1245307 | static | -3.5988 | -3.6949 | 12.39 | 12.5728 | 12.6831 | 92.4 | 94.4 | 91.6 |
| mp-1245067 | static | -3.5979 | -3.694 | 12.187 | 12.7035 | 12.8541 | 93.4 | 96.8 | 93.3 |
| oqmd-1214770 | static | -3.5957 | -3.6917 | 10.2896 | 10.2896 | 10.2896 | 90.0 | 90.0 | 90.0 |
| oqmd-1214859 | dynamic | -3.5932 | -3.6893 | 7.1561 | 7.1561 | 7.1561 | 90.0 | 90.0 | 90.0 |
| oqmd-1214859 | box | -3.5929 | -3.6889 | 7.1596 | 7.1596 | 7.1596 | 90.0 | 90.0 | 90.0 |
| mp-1245129 | box | -3.5911 | -3.6872 | 12.1311 | 12.5238 | 12.814 | 93.6 | 92.1 | 96.8 |
| mp-1245152 | box | -3.59 | -3.6861 | 12.3149 | 12.4722 | 12.6996 | 91.8 | 91.9 | 95.7 |
| oqmd-1214770 | box | -3.5893 | -3.6854 | 10.2339 | 10.2339 | 10.2339 | 90.0 | 90.0 | 90.0 |
| mp-1244953 | box | -3.5893 | -3.6853 | 12.4368 | 12.4937 | 12.6764 | 85.6 | 80.5 | 87.1 |
| A5--beta-Sn | static | -3.5886 | -3.6846 | 5.4909 | 5.4909 | 2.7866 | 90.0 | 90.0 | 90.0 |
| mp-1245307 | box | -3.5838 | -3.6799 | 12.425 | 12.5417 | 12.559 | 90.6 | 92.4 | 94.1 |
| mp-1245067 | box | -3.5829 | -3.6789 | 12.3667 | 12.6829 | 12.688 | 92.7 | 97.6 | 93.8 |
| Ah--alpha-Po--sc | dynamic | -3.5736 | -3.6696 | 2.8473 | 2.8473 | 2.8473 | 90.0 | 90.0 | 90.0 |
| A15--beta-W | static | -3.5476 | -3.6437 | 5.3498 | 5.3498 | 5.3498 | 90.0 | 90.0 | 90.0 |
| oqmd-1214681 | box | -3.5187 | -3.6148 | 4.5879 | 8.4325 | 4.6579 | 90.0 | 90.0 | 90.0 |
| oqmd-1215928 | box | -3.4167 | -3.5127 | 4.6743 | 4.6743 | 5.5422 | 90.0 | 90.0 | 120.0 |
| mp-1239196 | box | -3.3656 | -3.4616 | 3.8755 | 3.8755 | 14.0321 | 90.0 | 90.0 | 90.0 |
| A4--C--dc | static | -3.3278 | -3.4239 | 6.127 | 6.127 | 6.127 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-16511 | dynamic | -5.1146 | -5.1885 | 7.5156 | 7.5156 | 7.5932 | 90.0 | 90.0 | 120.0 |
| mp-16511 | static | -5.1146 | -5.1885 | 7.5157 | 7.5157 | 7.5931 | 90.0 | 90.0 | 120.0 |
| mp-16511 | box | -5.1102 | -5.184 | 7.5322 | 7.5322 | 7.5677 | 90.0 | 90.0 | 120.0 |
| oqmd-10499 | box | -5.1063 | -5.1801 | 7.5567 | 7.5567 | 7.5345 | 90.0 | 90.0 | 120.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-2856 | static | -5.3221 | -5.3925 | 4.9895 | 5.0393 | 8.7224 | 74.1 | 86.3 | 79.7 |
| oqmd-2782 | static | -5.3221 | -5.3925 | 4.9895 | 5.0396 | 8.7218 | 74.1 | 86.3 | 79.7 |
| mp-2856 | box | -5.3108 | -5.3812 | 4.9493 | 5.0363 | 8.7065 | 74.2 | 88.5 | 81.3 |
| oqmd-2782 | box | -5.3028 | -5.3732 | 4.9843 | 5.0175 | 8.6837 | 105.4 | 91.1 | 98.6 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| oqmd-27878 | dynamic | -4.1106 | -4.1993 | 7.5325 | 7.5325 | 7.5325 | 90.0 | 90.0 | 90.0 |
| mp-1104165 | box | -4.1042 | -4.1929 | 7.5275 | 7.5275 | 7.5275 | 90.0 | 90.0 | 90.0 |
| oqmd-27878 | box | -4.1031 | -4.1918 | 7.527 | 7.527 | 7.527 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-30526 | dynamic | -4.7511 | -4.8304 | 12.6603 | 12.6603 | 12.6603 | 90.0 | 90.0 | 90.0 |
| mp-30526 | box | -4.7436 | -4.823 | 12.6771 | 12.6771 | 12.6771 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-1498 | dynamic | -4.6204 | -4.6911 | 12.9749 | 12.9749 | 10.9191 | 90.0 | 90.0 | 90.0 |
| mp-1498 | static | -4.6186 | -4.6893 | 13.1139 | 13.1139 | 10.7986 | 90.0 | 90.0 | 90.0 |
| mp-1498 | box | -4.6141 | -4.6848 | 13.1717 | 13.1717 | 10.817 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
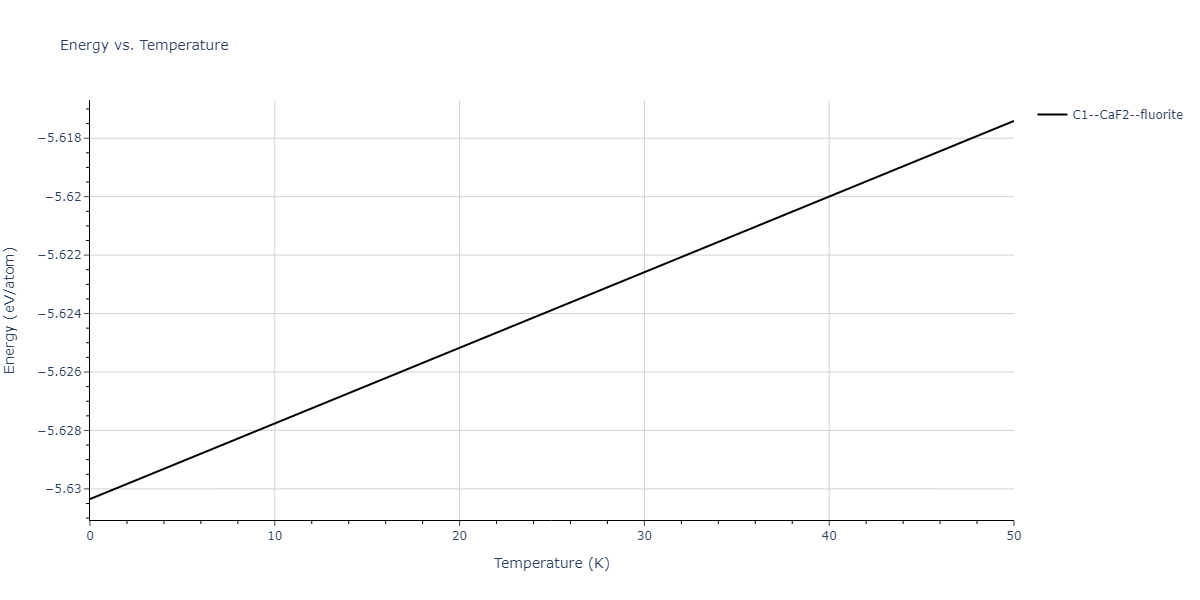
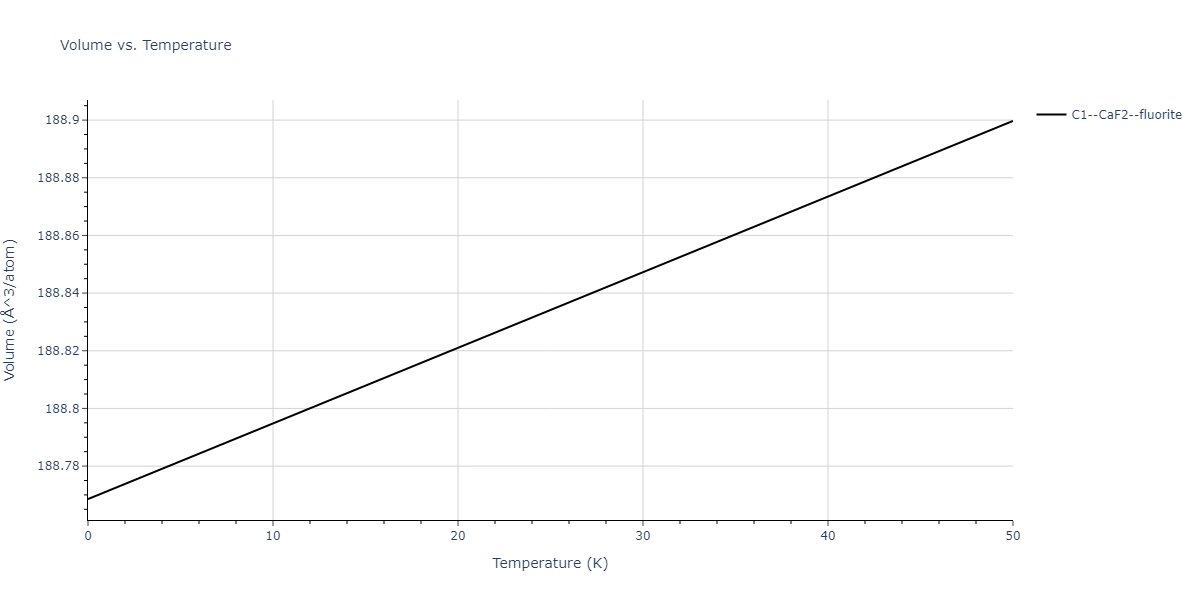
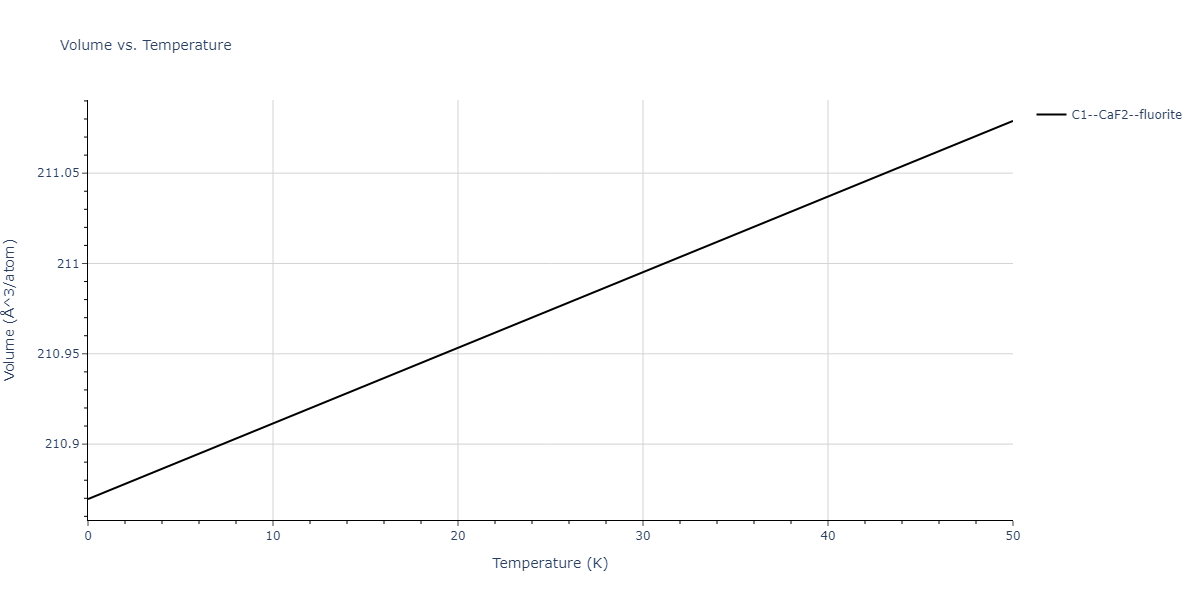
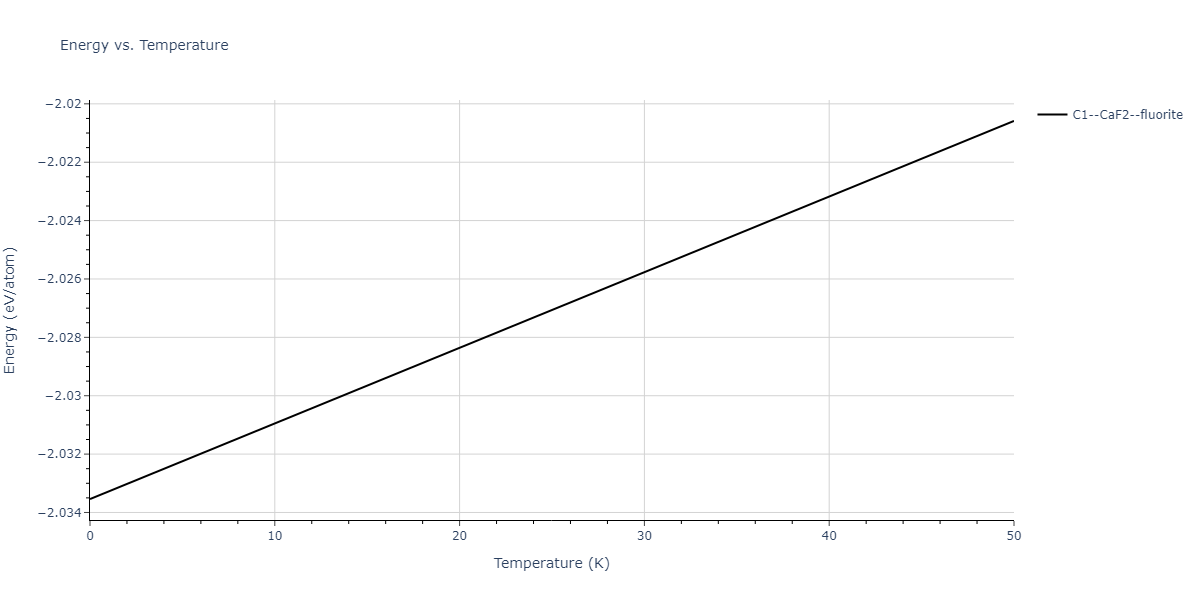
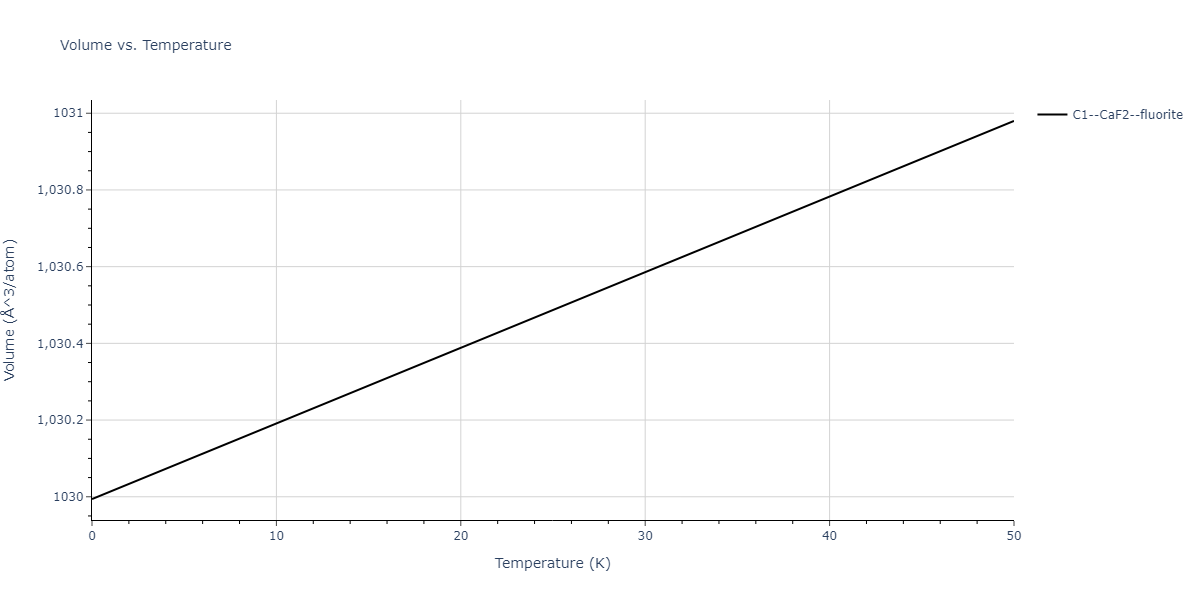
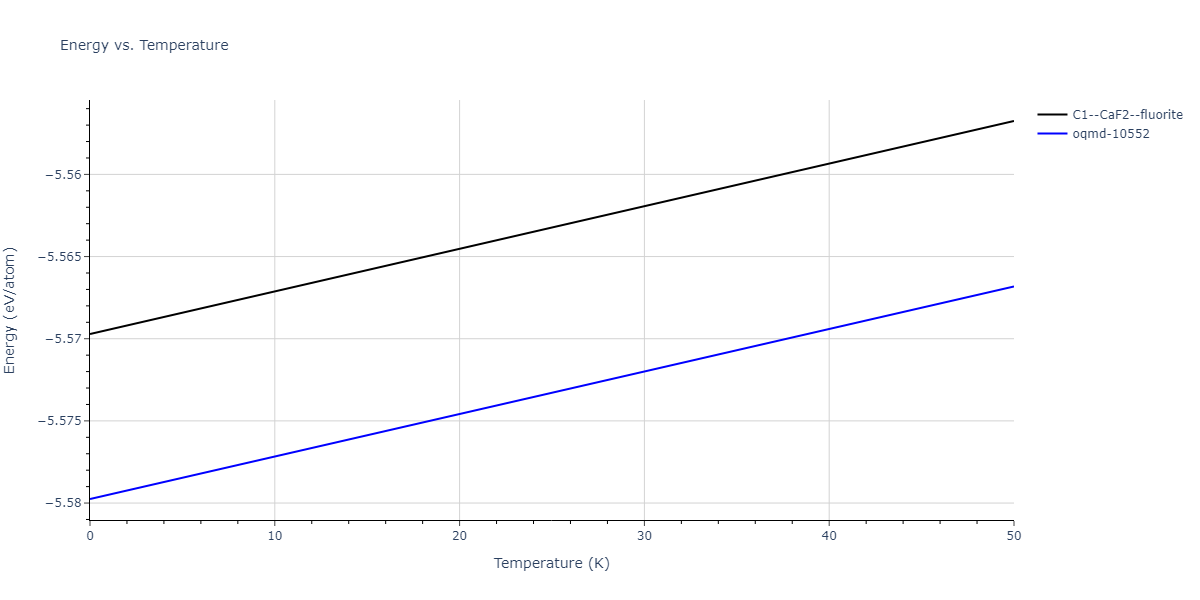
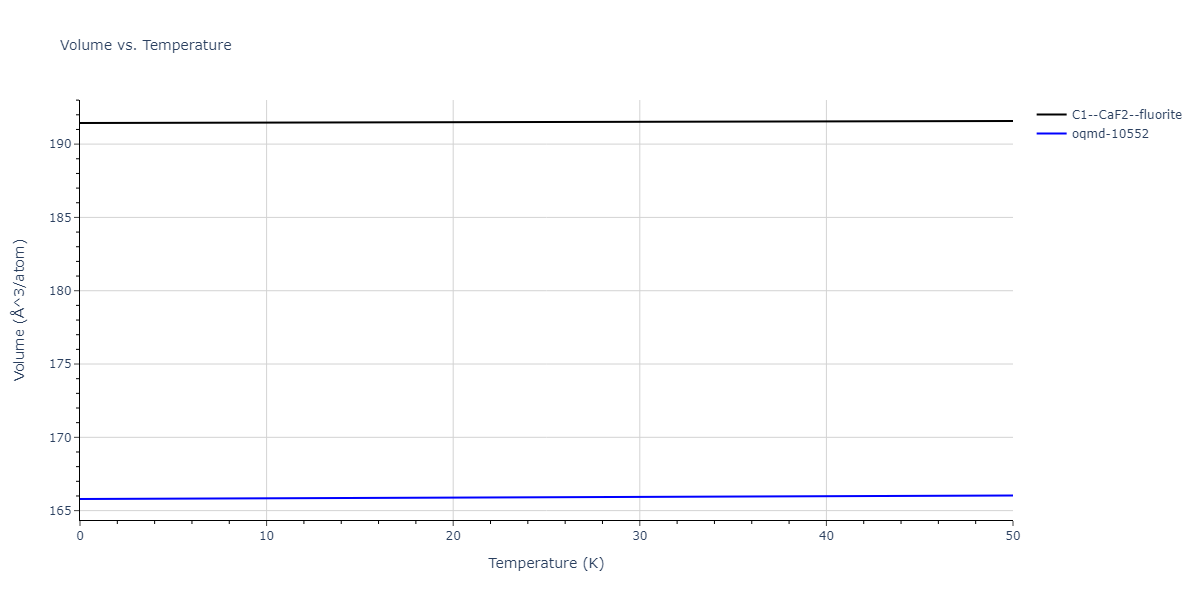
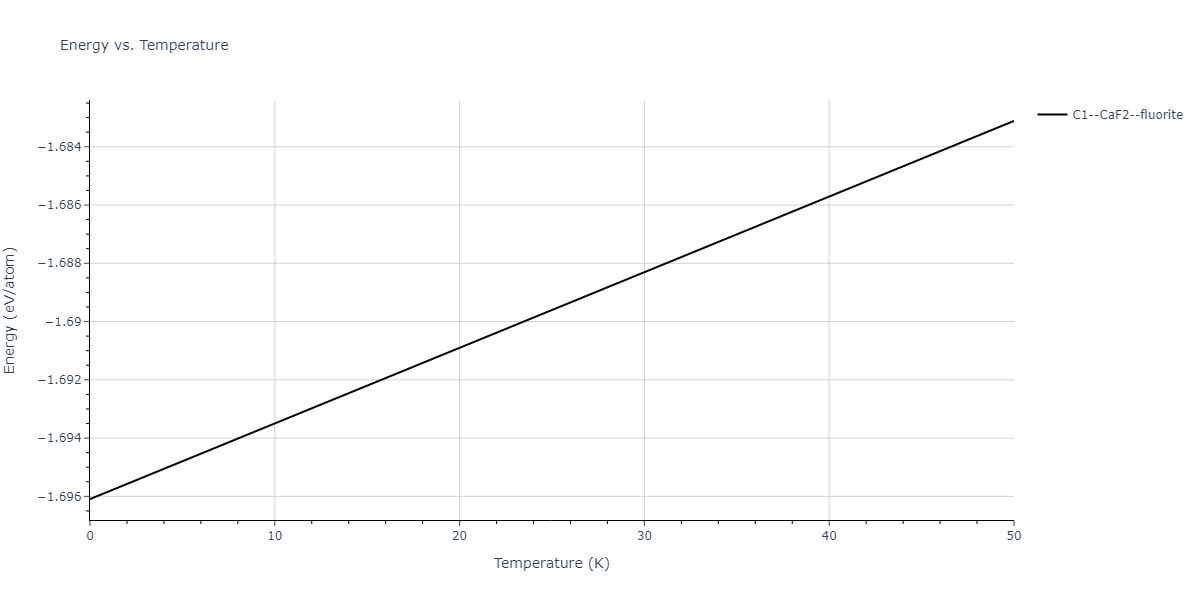
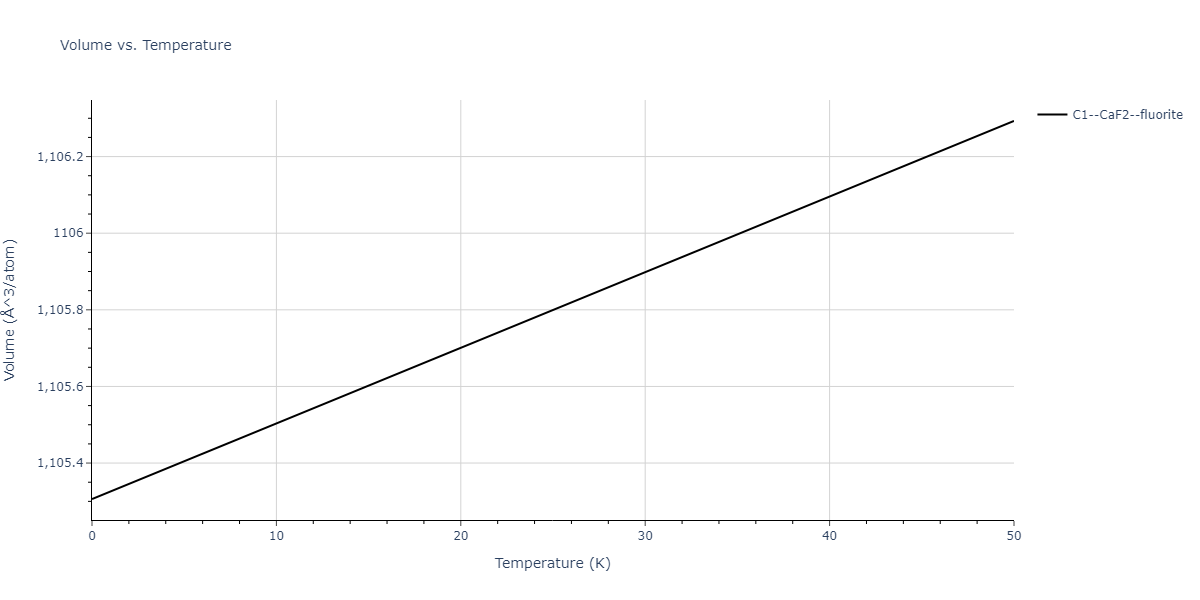
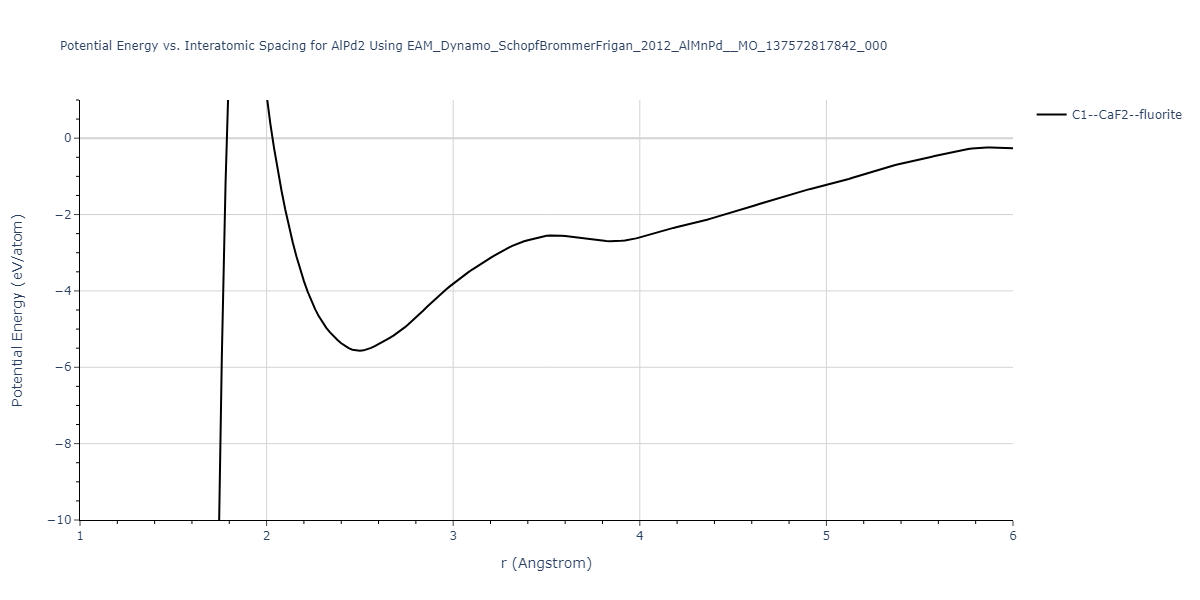
| C1--CaF2--fluorite | dynamic | -5.5664 | -5.6303 | 5.7365 | 5.7365 | 5.7365 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | static | -1.9426 | -2.0066 | 10.0686 | 10.0686 | 10.0686 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
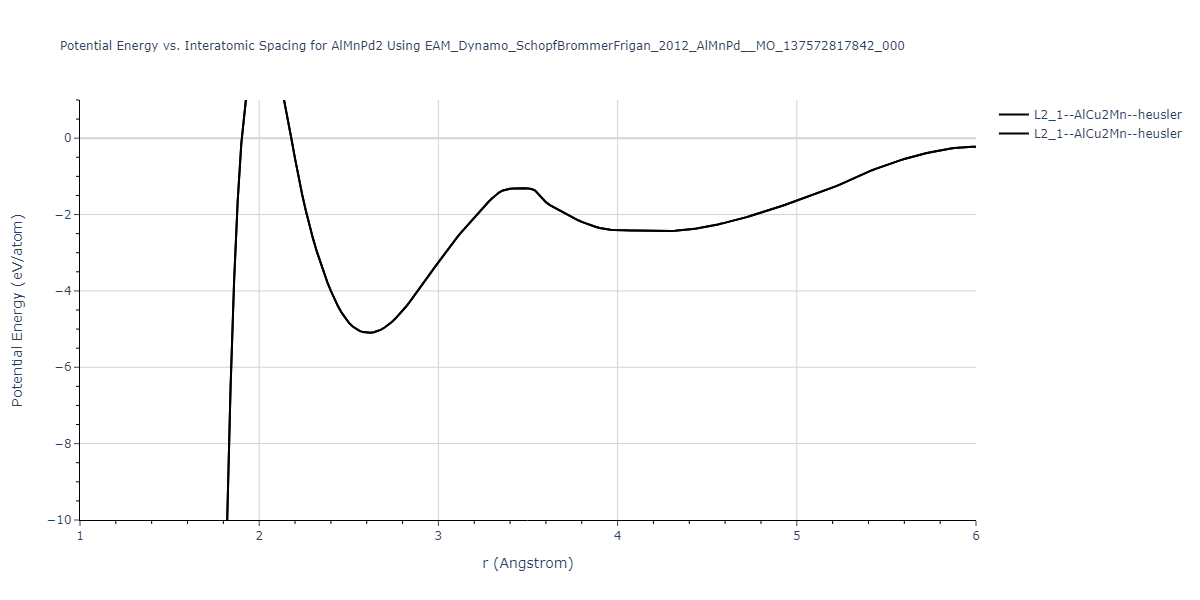
L2_1--AlCu2Mn--heusler = oqmd-510324
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| L2_1--AlCu2Mn--heusler | dynamic | -5.665 | -5.714 | 5.9264 | 5.9264 | 5.9264 | 90.0 | 90.0 | 90.0 |
| L2_1--AlCu2Mn--heusler | static | -2.7495 | -2.7985 | 9.3882 | 9.3882 | 9.3882 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
C1--CaF2--fluorite = mp-16522, oqmd-10553
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| C1--CaF2--fluorite | dynamic | -4.8129 | -4.8783 | 5.9521 | 5.9521 | 5.9521 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
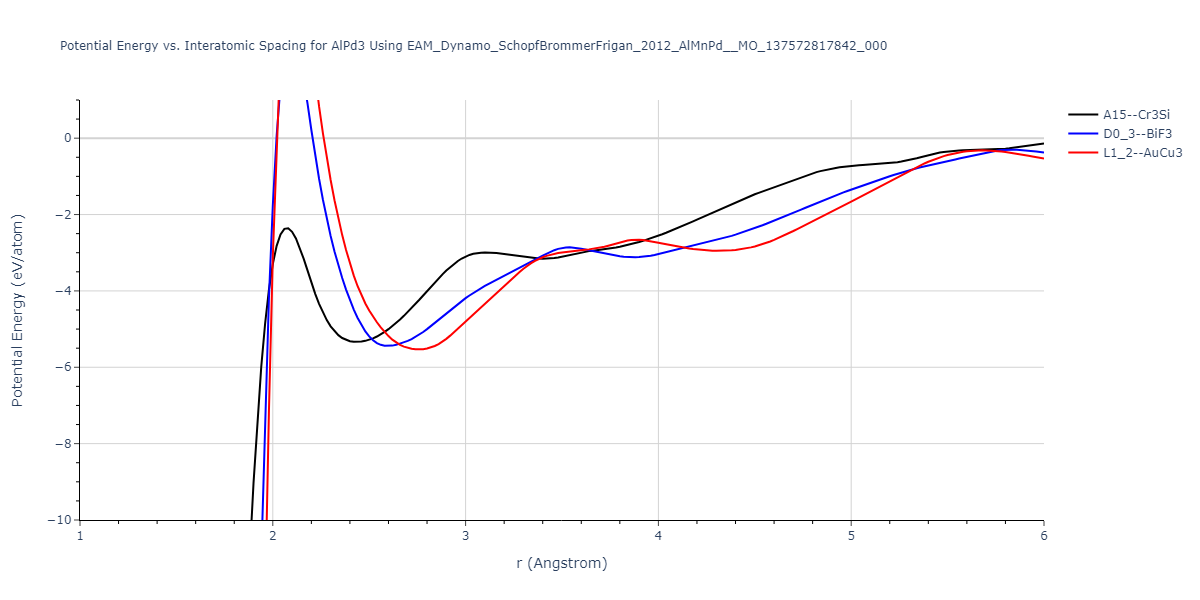
D0_3--BiF3 = oqmd-309860
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| L1_2--AuCu3 | dynamic | -5.1339 | -5.2059 | 3.7678 | 3.7678 | 3.7678 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | dynamic | -5.1127 | -5.1847 | 5.9027 | 5.9027 | 5.9027 | 90.0 | 90.0 | 90.0 |
| oqmd-299318 | static | -5.106 | -5.178 | 3.7262 | 3.7262 | 7.82 | 90.0 | 90.0 | 90.0 |
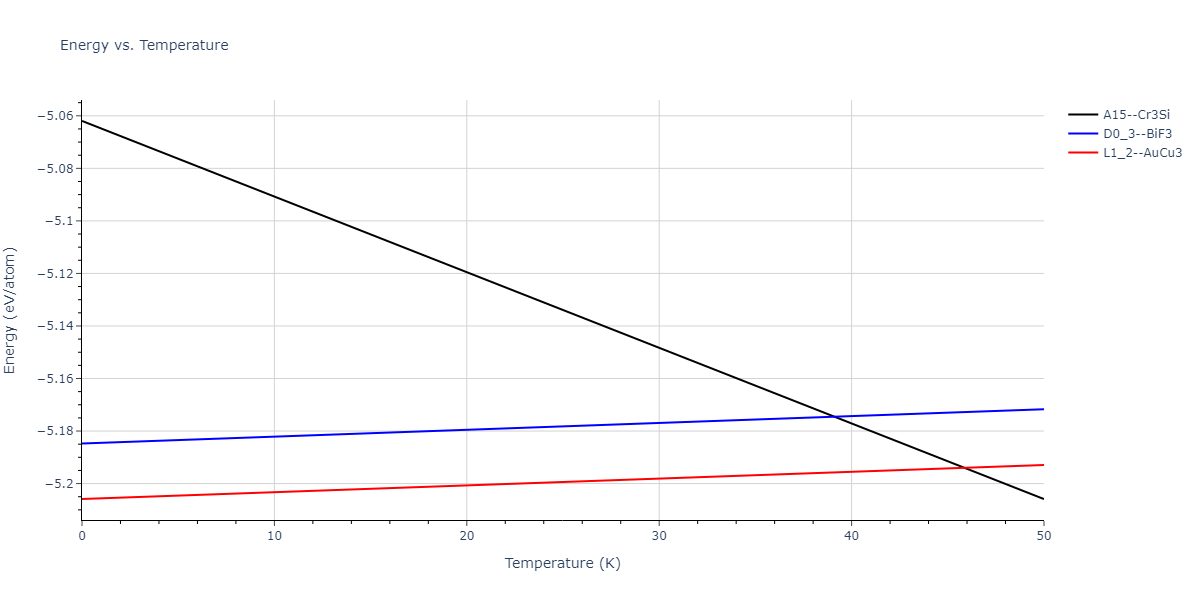
| A15--Cr3Si | dynamic | -4.99 | -5.062 | 4.8086 | 4.8086 | 4.8086 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | static | -2.6904 | -2.7623 | 7.3052 | 7.3052 | 7.3052 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
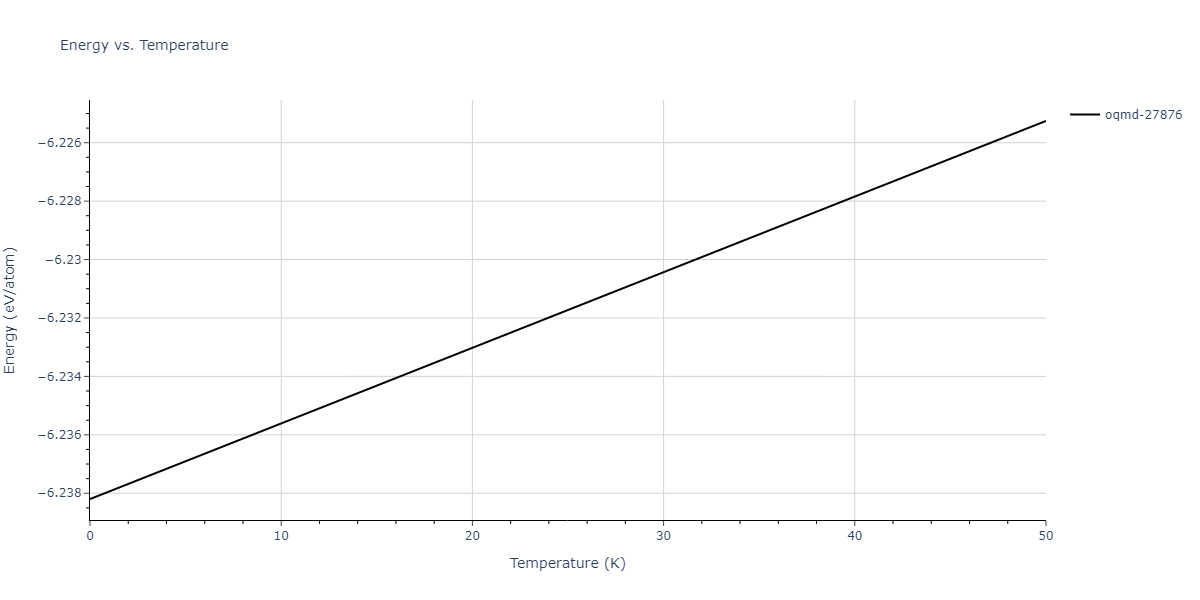
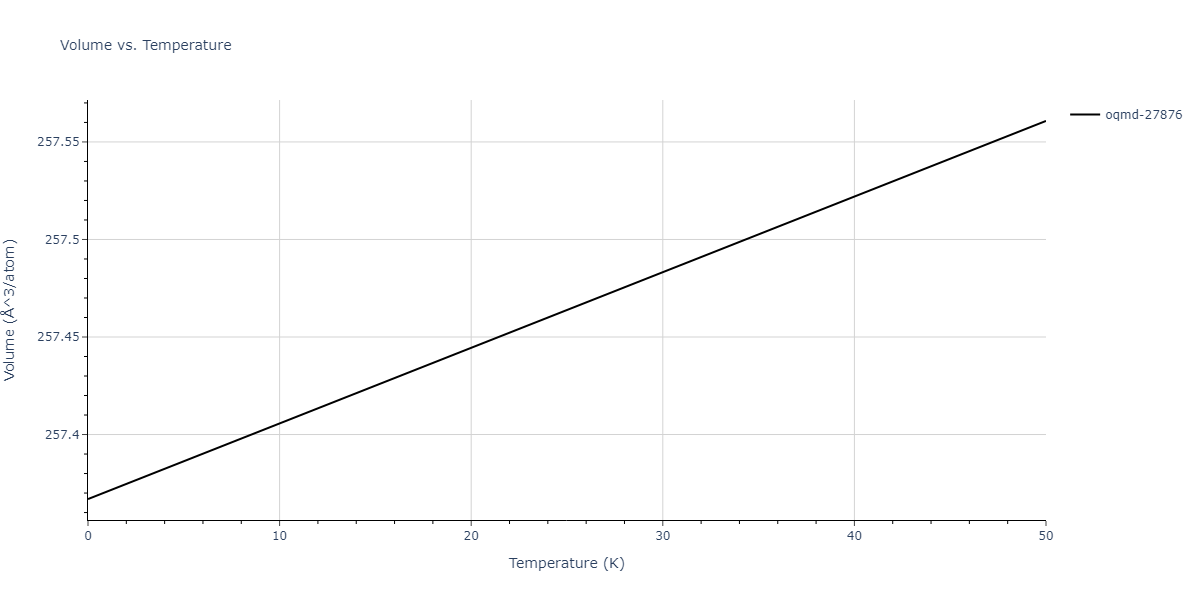
| oqmd-27876 | dynamic | -6.1806 | -6.2382 | 6.3609 | 6.3609 | 6.3609 | 90.0 | 90.0 | 90.0 |
| oqmd-27876 | box | -6.1513 | -6.2089 | 6.3639 | 6.3639 | 6.3639 | 90.0 | 90.0 | 90.0 |
| mp-1180309 | box | -6.15 | -6.2076 | 6.3638 | 6.3638 | 6.3638 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
D0_3--BiF3 = oqmd-309009
L1_2--AuCu3 = mp-1183127
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| L1_2--AuCu3 | dynamic | -4.5042 | -4.5773 | 3.954 | 3.954 | 3.954 | 90.0 | 90.0 | 90.0 |
| oqmd-298471 | dynamic | -4.4882 | -4.5612 | 3.9453 | 3.9453 | 7.9471 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | static | -4.4668 | -4.5399 | 6.2185 | 6.2185 | 6.2185 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | static | -4.4311 | -4.5042 | 5.0115 | 5.0115 | 5.0115 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-10901 | dynamic | -5.038 | -5.0973 | 4.2079 | 4.2079 | 5.0699 | 90.0 | 90.0 | 120.0 |
| mp-10901 | box | -5.0327 | -5.092 | 4.211 | 4.211 | 5.0729 | 90.0 | 90.0 | 120.0 |
| oqmd-10554 | box | -5.0326 | -5.0919 | 4.2099 | 4.2099 | 5.075 | 90.0 | 90.0 | 120.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-16523 | dynamic | -5.5257 | -5.5643 | 5.3078 | 10.1047 | 4.0835 | 90.0 | 90.0 | 90.0 |
| mp-16523 | static | -5.5257 | -5.5643 | 5.3078 | 10.1049 | 4.0834 | 90.0 | 90.0 | 90.0 |
| mp-16523 | box | -5.5142 | -5.5528 | 5.3307 | 10.2896 | 4.0467 | 90.0 | 90.0 | 90.0 |
| oqmd-10555 | box | -5.5136 | -5.5522 | 5.3327 | 10.3023 | 4.044 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| oqmd-10498 | static | -4.5423 | -4.6246 | 7.5836 | 6.658 | 8.5581 | 90.0 | 90.0 | 90.0 |
| mp-173 | static | -4.5423 | -4.6246 | 7.5836 | 6.6581 | 8.5582 | 90.0 | 90.0 | 90.0 |
| mp-173 | box | -4.5385 | -4.6208 | 7.5925 | 6.5817 | 8.6826 | 90.0 | 90.0 | 90.0 |
| oqmd-10498 | box | -4.5374 | -4.6197 | 7.5908 | 6.5837 | 8.6907 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-1194040 | dynamic | -5.9005 | -5.9596 | 12.5936 | 12.5936 | 7.4251 | 90.0 | 90.0 | 120.0 |
| mp-1194040 | static | -5.9005 | -5.9596 | 12.5938 | 12.5938 | 7.4257 | 90.0 | 90.0 | 120.0 |
| mp-1194040 | box | -5.8714 | -5.9304 | 12.6685 | 12.6685 | 7.5097 | 90.0 | 90.0 | 120.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-1212295 | dynamic | -5.5419 | -5.6084 | 14.6396 | 12.1566 | 12.2606 | 90.0 | 90.0 | 90.0 |
| mp-1212295 | static | -5.5419 | -5.6084 | 14.6415 | 12.156 | 12.2589 | 90.0 | 90.0 | 90.0 |
| mp-1212295 | box | -5.5072 | -5.5737 | 14.5583 | 12.0709 | 12.2594 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
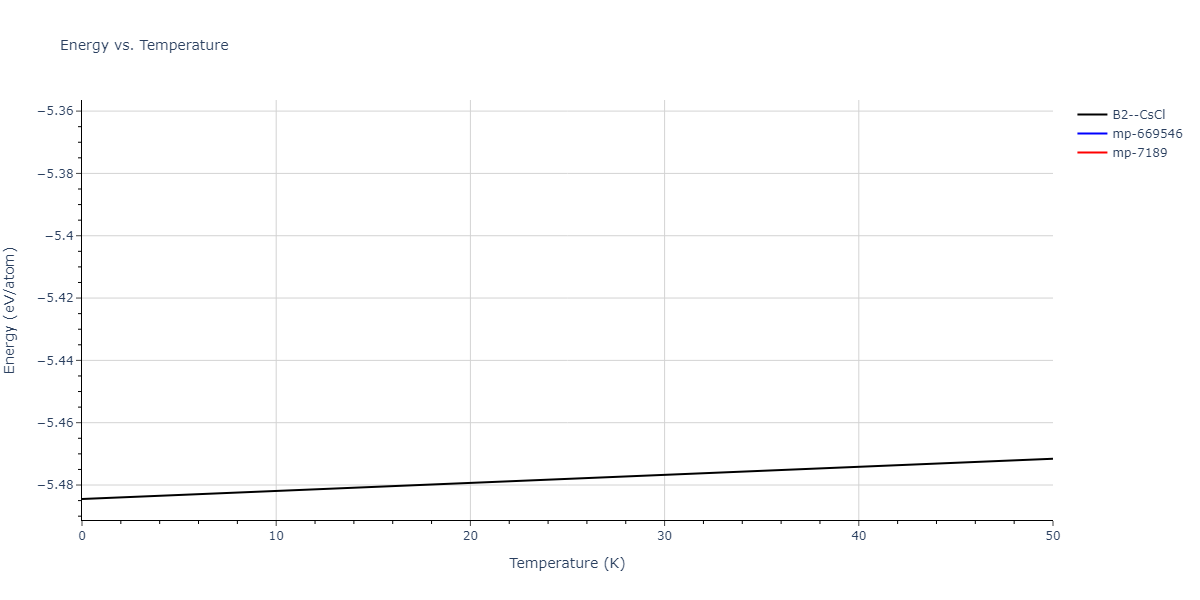
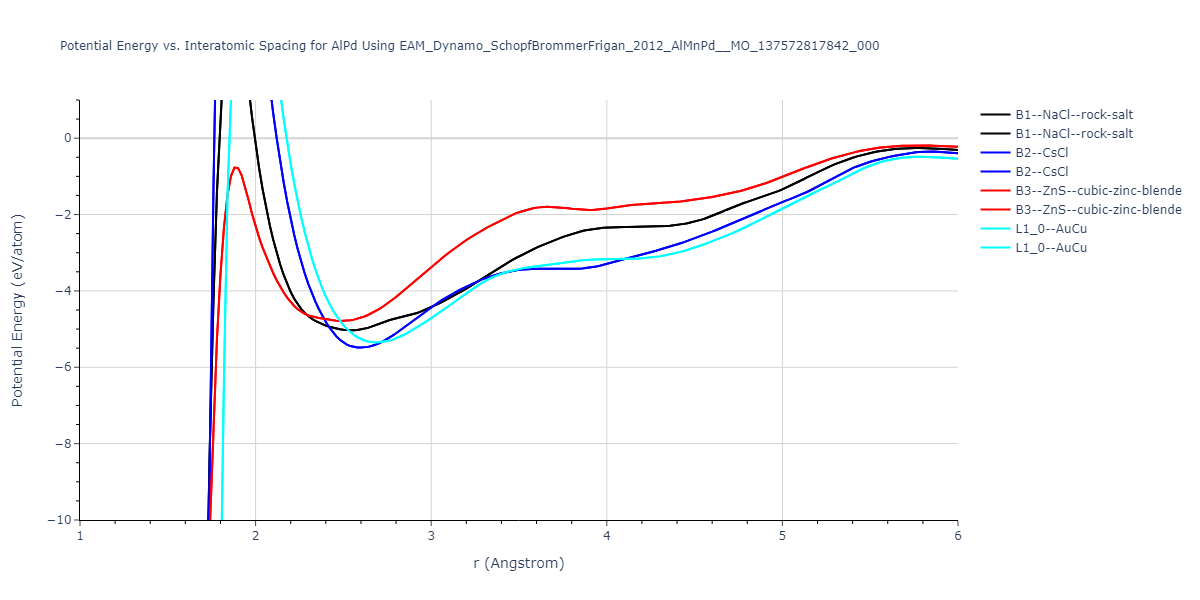
B2--CsCl = mp-12067, oqmd-658675
L1_0--AuCu = mp-771, oqmd-27877
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| B2--CsCl | dynamic | -6.8176 | -6.8656 | 2.838 | 2.838 | 2.838 | 90.0 | 90.0 | 90.0 |
| B1--NaCl--rock-salt | static | -5.8 | -5.8479 | 4.4032 | 4.4032 | 4.4032 | 90.0 | 90.0 | 90.0 |
| B3--ZnS--cubic-zinc-blende | static | -4.6459 | -4.6939 | 5.756 | 5.756 | 5.756 | 90.0 | 90.0 | 90.0 |
| B2--CsCl | dynamic | -2.6194 | -2.6674 | 4.9991 | 4.9991 | 4.9991 | 90.0 | 90.0 | 90.0 |
| B1--NaCl--rock-salt | static | -1.6171 | -1.665 | 9.1482 | 9.1482 | 9.1482 | 90.0 | 90.0 | 90.0 |
| B3--ZnS--cubic-zinc-blende | dynamic | -1.2671 | -1.3151 | 10.6946 | 10.6946 | 10.6946 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| C1--CaF2--fluorite | static | -6.3298 | -6.3617 | 5.1671 | 5.1671 | 5.1671 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | dynamic | -2.0016 | -2.0335 | 10.099 | 10.099 | 10.099 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | static | -1.5223 | -1.5542 | 9.5609 | 9.5609 | 9.5609 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
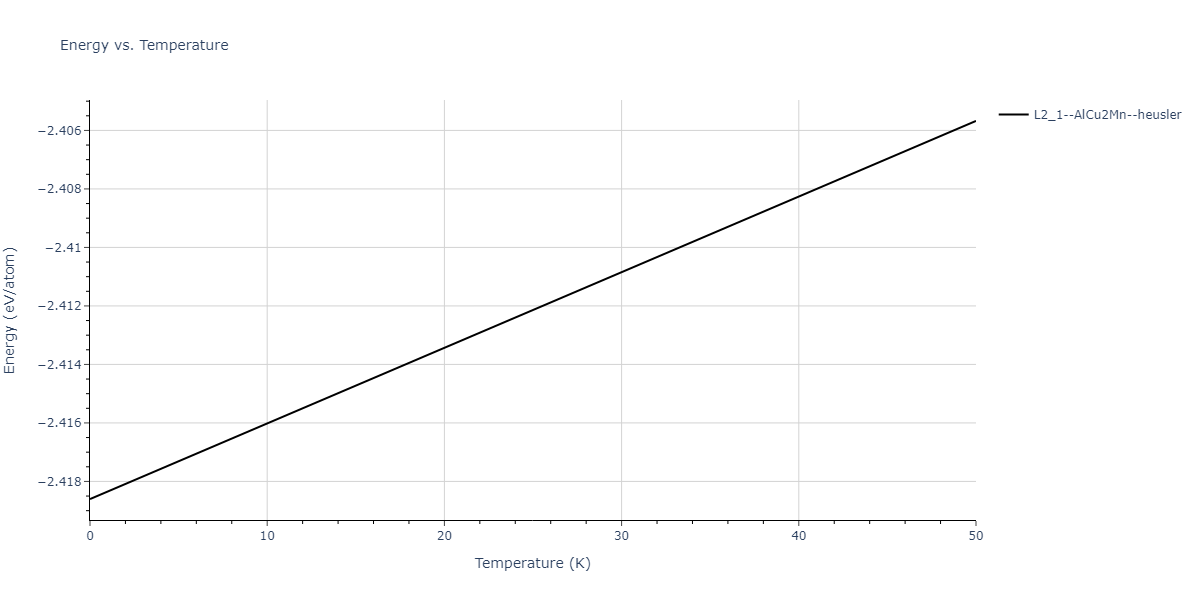
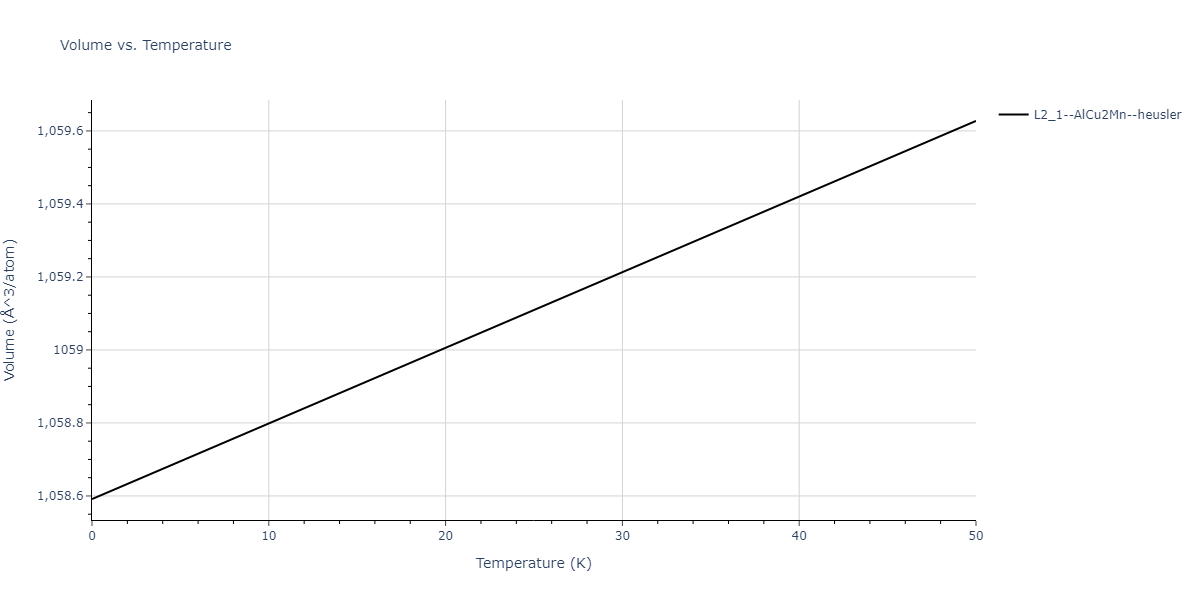
L2_1--AlCu2Mn--heusler = oqmd-471923
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| L2_1--AlCu2Mn--heusler | static | -5.3173 | -5.3423 | 5.9156 | 5.9156 | 5.9156 | 90.0 | 90.0 | 90.0 |
| L2_1--AlCu2Mn--heusler | dynamic | -2.3936 | -2.4186 | 10.1916 | 10.1916 | 10.1916 | 90.0 | 90.0 | 90.0 |
| L2_1--AlCu2Mn--heusler | static | -0.9251 | -0.95 | 7.7596 | 7.7596 | 7.7596 | 90.0 | 90.0 | 90.0 |
| L2_1--AlCu2Mn--heusler | box | -0.9134 | -0.9384 | 7.9865 | 7.9865 | 7.9865 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
D0_3--BiF3 = mp-973149, oqmd-22372
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| D0_3--BiF3 | dynamic | -8.1364 | -8.1603 | 5.6328 | 5.6328 | 5.6328 | 90.0 | 90.0 | 90.0 |
| L1_2--AuCu3 | dynamic | -8.0957 | -8.1196 | 3.5115 | 3.5115 | 3.5115 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | static | -7.9362 | -7.9601 | 4.4649 | 4.4649 | 4.4649 | 90.0 | 90.0 | 90.0 |
| oqmd-299450 | box | -6.2926 | -6.3165 | 3.072 | 3.072 | 12.0723 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | dynamic | -2.48 | -2.5039 | 8.083 | 8.083 | 8.083 | 90.0 | 90.0 | 90.0 |
| L1_2--AuCu3 | dynamic | -2.4684 | -2.4923 | 6.6705 | 6.6705 | 6.6705 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | dynamic | -2.385 | -2.4089 | 10.301 | 10.301 | 10.301 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
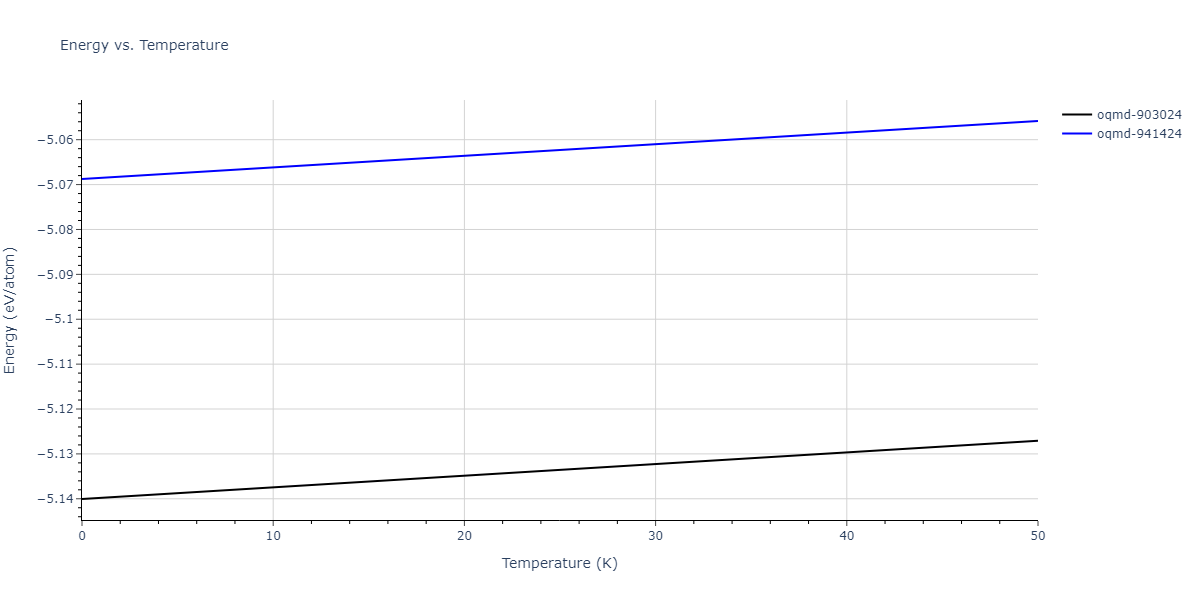
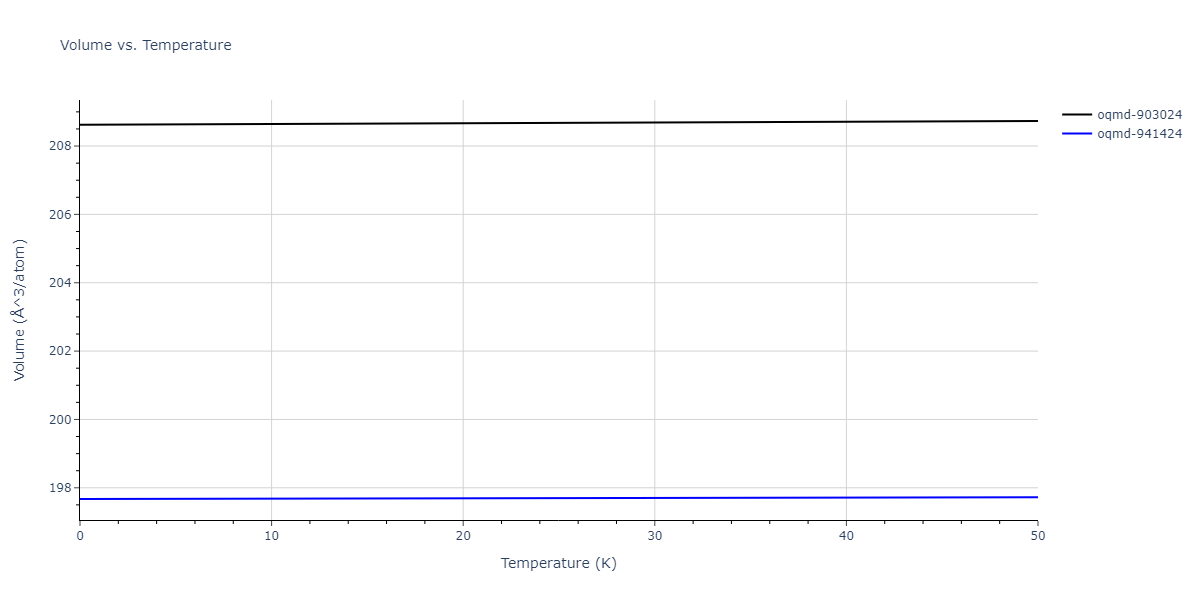
| oqmd-903024 | dynamic | -5.1067 | -5.14 | 5.9309 | 5.9309 | 5.9309 | 90.0 | 90.0 | 90.0 |
| oqmd-941424 | dynamic | -5.0354 | -5.0688 | 5.8253 | 5.8253 | 5.8253 | 90.0 | 90.0 | 90.0 |
| oqmd-898211 | static | -4.7193 | -4.7526 | 5.9098 | 5.9098 | 5.9098 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
L2_1--AlCu2Mn--heusler = mp-10891, oqmd-10501
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| L2_1--AlCu2Mn--heusler | static | -5.0706 | -5.0967 | 6.0301 | 6.0301 | 6.0301 | 90.0 | 90.0 | 90.0 |
| L2_1--AlCu2Mn--heusler | static | -2.4097 | -2.4358 | 9.7906 | 9.7906 | 9.7906 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
B2--CsCl = mp-829, oqmd-10550
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| B2--CsCl | dynamic | -5.4344 | -5.4845 | 2.9988 | 2.9988 | 2.9988 | 90.0 | 90.0 | 90.0 |
| mp-7189 | dynamic | -5.3263 | -5.3764 | 4.855 | 4.855 | 4.855 | 90.0 | 90.0 | 90.0 |
| oqmd-10551 | box | -5.3259 | -5.3759 | 4.8544 | 4.8544 | 4.8544 | 90.0 | 90.0 | 90.0 |
| mp-7189 | box | -5.3258 | -5.3758 | 4.8541 | 4.8541 | 4.8541 | 90.0 | 90.0 | 90.0 |
| mp-669546 | dynamic | -5.3131 | -5.3632 | 15.6172 | 15.6172 | 5.2613 | 90.0 | 90.0 | 120.0 |
| mp-669546 | static | -5.3131 | -5.3632 | 15.6167 | 15.6167 | 5.2615 | 90.0 | 90.0 | 120.0 |
| oqmd-10549 | static | -5.3103 | -5.3604 | 15.6007 | 15.6007 | 5.2661 | 90.0 | 90.0 | 120.0 |
| mp-669546 | box | -5.3089 | -5.359 | 15.5958 | 15.5958 | 5.2675 | 90.0 | 90.0 | 120.0 |
| oqmd-10549 | box | -5.3089 | -5.3589 | 15.5969 | 15.5969 | 5.2673 | 90.0 | 90.0 | 120.0 |
| B1--NaCl--rock-salt | static | -4.9838 | -5.0338 | 5.0935 | 5.0935 | 5.0935 | 90.0 | 90.0 | 90.0 |
| B3--ZnS--cubic-zinc-blende | static | -4.7358 | -4.7859 | 5.7466 | 5.7466 | 5.7466 | 90.0 | 90.0 | 90.0 |
| B2--CsCl | static | -3.3785 | -3.4286 | 4.3399 | 4.3399 | 4.3399 | 90.0 | 90.0 | 90.0 |
| B3--ZnS--cubic-zinc-blende | static | -1.8291 | -1.8792 | 9.0083 | 9.0083 | 9.0083 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| oqmd-10552 | dynamic | -5.545 | -5.5798 | 5.3084 | 4.0593 | 7.694 | 90.0 | 90.0 | 90.0 |
| mp-2824 | static | -5.545 | -5.5798 | 5.3084 | 4.0593 | 7.6942 | 90.0 | 90.0 | 90.0 |
| oqmd-10552 | box | -5.539 | -5.5738 | 5.3575 | 4.061 | 7.6617 | 90.0 | 90.0 | 90.0 |
| mp-2824 | box | -5.5387 | -5.5734 | 5.3567 | 4.0593 | 7.669 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | dynamic | -5.5349 | -5.5697 | 5.7635 | 5.7635 | 5.7635 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | static | -2.6637 | -2.6985 | 8.8964 | 8.8964 | 8.8964 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
D0_3--BiF3 = oqmd-309410
L1_2--AuCu3 = mp-1183143
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| L1_2--AuCu3 | dynamic | -5.5047 | -5.5318 | 3.8973 | 3.8973 | 3.8973 | 90.0 | 90.0 | 90.0 |
| oqmd-298869 | dynamic | -5.4865 | -5.5136 | 3.9012 | 3.9012 | 7.7909 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | static | -5.415 | -5.4421 | 5.997 | 5.997 | 5.997 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | static | -5.3095 | -5.3366 | 4.8663 | 4.8663 | 4.8663 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | static | -3.1315 | -3.1586 | 6.8171 | 6.8171 | 6.8171 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | static | -3.0896 | -3.1167 | 8.9486 | 8.9486 | 8.9486 | 90.0 | 90.0 | 90.0 |
| L1_2--AuCu3 | dynamic | -2.9234 | -2.9505 | 6.1018 | 6.1018 | 6.1018 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-605663 | dynamic | -5.4484 | -5.4679 | 10.9908 | 3.9281 | 8.1999 | 90.0 | 90.0 | 90.0 |
| oqmd-23084 | static | -5.4484 | -5.4679 | 10.9904 | 3.9282 | 8.1996 | 90.0 | 90.0 | 90.0 |
| mp-605663 | box | -5.4345 | -5.454 | 10.7697 | 3.9763 | 8.0713 | 90.0 | 90.0 | 90.0 |
| oqmd-23084 | box | -5.4334 | -5.4528 | 10.7539 | 3.979 | 8.0636 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
A1--Cu--fcc = mp-8634, oqmd-676149
A2--W--bcc = mp-1055908
A5--beta-Sn = mp-1057139
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
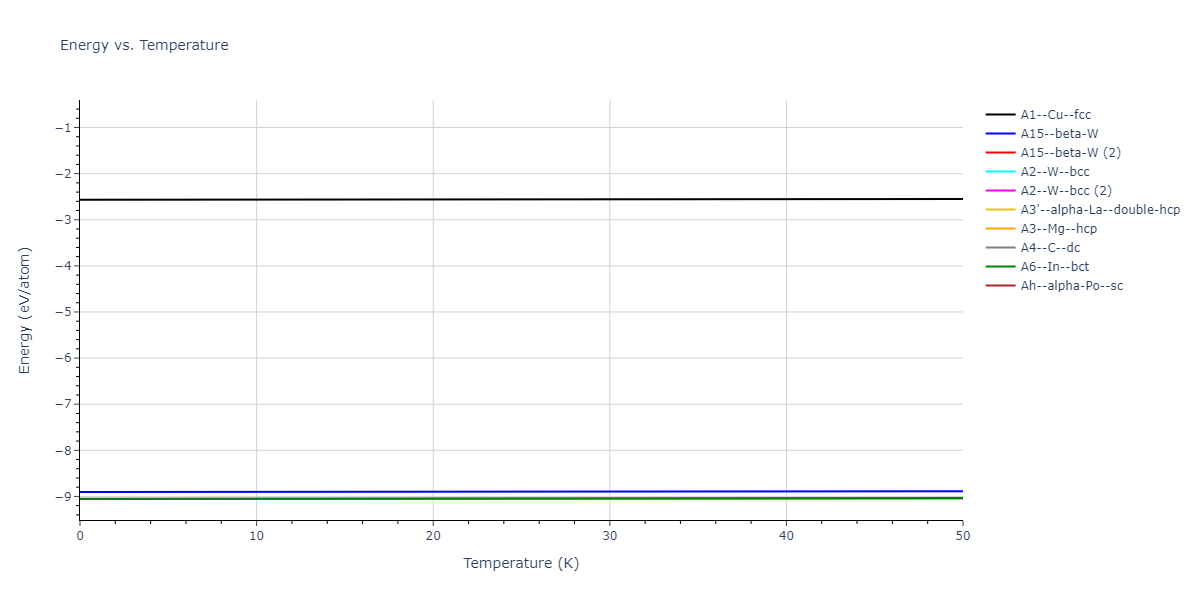
| A2--W--bcc | dynamic | -9.0564 | -9.0562 | 2.788 | 2.788 | 2.788 | 90.0 | 90.0 | 90.0 |
| A6--In--bct | dynamic | -9.0493 | -9.0491 | 2.3146 | 2.3146 | 3.9688 | 90.0 | 90.0 | 90.0 |
| A1--Cu--fcc | static | -9.0075 | -9.0074 | 3.4746 | 3.4746 | 3.4746 | 90.0 | 90.0 | 90.0 |
| A3'--alpha-La--double-hcp | static | -9.0008 | -9.0006 | 2.4518 | 2.4518 | 8.103 | 90.0 | 90.0 | 120.0 |
| A3--Mg--hcp | static | -8.998 | -8.9978 | 2.4487 | 2.4487 | 4.088 | 90.0 | 90.0 | 120.0 |
| mp-35 | static | -8.9643 | -8.9641 | 8.5108 | 8.5108 | 8.5108 | 90.0 | 90.0 | 90.0 |
| oqmd-7874 | static | -8.9641 | -8.9639 | 8.5122 | 8.5122 | 8.5122 | 90.0 | 90.0 | 90.0 |
| oqmd-7874 | box | -8.9613 | -8.9612 | 8.5118 | 8.5118 | 8.5118 | 90.0 | 90.0 | 90.0 |
| mp-35 | box | -8.9588 | -8.9587 | 8.5049 | 8.5049 | 8.5049 | 90.0 | 90.0 | 90.0 |
| A5--beta-Sn | static | -8.9038 | -8.9036 | 4.2508 | 4.2508 | 2.3737 | 90.0 | 90.0 | 90.0 |
| A15--beta-W | dynamic | -8.9028 | -8.9027 | 4.3856 | 4.3856 | 4.3856 | 90.0 | 90.0 | 90.0 |
| mp-542909 | static | -8.8824 | -8.8822 | 6.0015 | 6.0015 | 6.0015 | 90.0 | 90.0 | 90.0 |
| mp-542909 | box | -8.8607 | -8.8605 | 5.9919 | 5.9919 | 5.9919 | 90.0 | 90.0 | 90.0 |
| oqmd-7587 | box | -8.8541 | -8.8539 | 5.9908 | 5.9908 | 5.9908 | 90.0 | 90.0 | 90.0 |
| Ah--alpha-Po--sc | static | -8.6352 | -8.6351 | 2.1918 | 2.1918 | 2.1918 | 90.0 | 90.0 | 90.0 |
| A7--alpha-As | box | -8.292 | -8.2919 | 2.9524 | 2.9524 | 8.5604 | 90.0 | 90.0 | 120.0 |
| mp-1191484 | static | -8.0655 | -8.0654 | 10.2722 | 10.2722 | 10.2722 | 90.0 | 90.0 | 90.0 |
| A4--C--dc | static | -7.0915 | -7.0914 | 4.447 | 4.447 | 4.447 | 90.0 | 90.0 | 90.0 |
| A3'--alpha-La--double-hcp | dynamic | -2.5654 | -2.5652 | 4.813 | 4.813 | 15.7191 | 90.0 | 90.0 | 120.0 |
| A3--Mg--hcp | dynamic | -2.5654 | -2.5652 | 4.813 | 4.813 | 7.8596 | 90.0 | 90.0 | 120.0 |
| A1--Cu--fcc | dynamic | -2.5654 | -2.5652 | 6.8066 | 6.8066 | 6.8066 | 90.0 | 90.0 | 90.0 |
| A15--beta-W | dynamic | -2.405 | -2.4049 | 8.3667 | 8.3667 | 8.3667 | 90.0 | 90.0 | 90.0 |
| A2--W--bcc | dynamic | -2.3933 | -2.3931 | 5.2715 | 5.2715 | 5.2715 | 90.0 | 90.0 | 90.0 |
| A5--beta-Sn | static | -1.433 | -1.4328 | 8.9846 | 8.9846 | 4.6398 | 90.0 | 90.0 | 90.0 |
| Ah--alpha-Po--sc | dynamic | -1.2943 | -1.2941 | 4.816 | 4.816 | 4.816 | 90.0 | 90.0 | 90.0 |
| A4--C--dc | dynamic | -0.8584 | -0.8583 | 11.1234 | 11.1234 | 11.1234 | 90.0 | 90.0 | 90.0 |
| A2--W--bcc | static | -0.8237 | -0.8235 | 4.0645 | 4.0645 | 4.0645 | 90.0 | 90.0 | 90.0 |
| A3--Mg--hcp | static | -0.8212 | -0.8211 | 3.62 | 3.62 | 5.9114 | 90.0 | 90.0 | 120.0 |
| A3'--alpha-La--double-hcp | static | -0.821 | -0.8209 | 3.62 | 3.62 | 11.8229 | 90.0 | 90.0 | 120.0 |
| A1--Cu--fcc | static | -0.821 | -0.8208 | 5.1477 | 5.1477 | 5.1477 | 90.0 | 90.0 | 90.0 |
| A6--In--bct | static | -0.8204 | -0.8203 | 3.56 | 3.56 | 5.34 | 90.0 | 90.0 | 90.0 |
| A2--W--bcc | box | -0.8129 | -0.8127 | 3.9533 | 3.9533 | 3.9533 | 90.0 | 90.0 | 90.0 |
| A3--Mg--hcp | box | -0.8118 | -0.8116 | 3.5278 | 3.5278 | 5.8047 | 90.0 | 90.0 | 120.0 |
| A3'--alpha-La--double-hcp | box | -0.8107 | -0.8106 | 3.5307 | 3.5307 | 11.5725 | 90.0 | 90.0 | 120.0 |
| A1--Cu--fcc | box | -0.8095 | -0.8093 | 4.9974 | 4.9974 | 4.9974 | 90.0 | 90.0 | 90.0 |
| A15--beta-W | static | -0.7914 | -0.7912 | 6.56 | 6.56 | 6.56 | 90.0 | 90.0 | 90.0 |
| A15--beta-W | box | -0.7758 | -0.7756 | 6.3489 | 6.3489 | 6.3489 | 90.0 | 90.0 | 90.0 |
| A4--C--dc | static | -0.2529 | -0.2528 | 8.7295 | 8.7295 | 8.7295 | 90.0 | 90.0 | 90.0 |
| A4--C--dc | box | -0.2497 | -0.2495 | 8.4638 | 8.4638 | 8.4638 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| C1--CaF2--fluorite | static | -3.3118 | -3.3131 | 5.9289 | 5.9289 | 5.9289 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | dynamic | -1.6948 | -1.6961 | 10.3394 | 10.3394 | 10.3394 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | static | -1.2032 | -1.2044 | 9.5147 | 9.5147 | 9.5147 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
D0_3--BiF3 = oqmd-309193
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| D0_3--BiF3 | static | -5.8672 | -5.8681 | 5.8796 | 5.8796 | 5.8796 | 90.0 | 90.0 | 90.0 |
| L1_2--AuCu3 | static | -5.7983 | -5.7993 | 3.6349 | 3.6349 | 3.6349 | 90.0 | 90.0 | 90.0 |
| oqmd-298655 | box | -5.6709 | -5.6718 | 3.7112 | 3.7112 | 7.1203 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | static | -5.6024 | -5.6033 | 4.663 | 4.663 | 4.663 | 90.0 | 90.0 | 90.0 |
| L1_2--AuCu3 | dynamic | -2.3971 | -2.398 | 6.7165 | 6.7165 | 6.7165 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | dynamic | -2.3214 | -2.3223 | 8.2175 | 8.2175 | 8.2175 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | dynamic | -2.2346 | -2.2355 | 10.4272 | 10.4272 | 10.4272 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | static | -0.6783 | -0.6792 | 8.1753 | 8.1753 | 8.1753 | 90.0 | 90.0 | 90.0 |
| L1_2--AuCu3 | static | -0.6361 | -0.637 | 5.4306 | 5.4306 | 5.4306 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | static | -0.5482 | -0.5491 | 6.68 | 6.68 | 6.68 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | box | -0.4941 | -0.495 | 6.32 | 6.32 | 6.32 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | box | -0.4844 | -0.4853 | 7.6984 | 7.6984 | 7.6984 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-1078895 | dynamic | -4.9498 | -4.9523 | 5.9957 | 8.6158 | 4.1472 | 90.0 | 90.0 | 90.0 |
| oqmd-684936 | static | -4.9498 | -4.9523 | 5.9956 | 8.6159 | 4.1472 | 90.0 | 90.0 | 90.0 |
| mp-1078895 | static | -4.9498 | -4.9523 | 5.9955 | 8.616 | 4.1472 | 90.0 | 90.0 | 90.0 |
| oqmd-684936 | box | -4.1684 | -4.1709 | 7.5631 | 8.1919 | 3.578 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
B2--CsCl = mp-1902, oqmd-90200
L1_0--AuCu = mp-238, oqmd-17676
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| L1_0--AuCu | static | -4.8026 | -4.8045 | 2.4667 | 2.4667 | 4.2779 | 90.0 | 90.0 | 90.0 |
| B2--CsCl | static | -3.9085 | -3.9104 | 3.0152 | 3.0152 | 3.0152 | 90.0 | 90.0 | 90.0 |
| B3--ZnS--cubic-zinc-blende | dynamic | -3.4157 | -3.4177 | 6.1235 | 6.1235 | 6.1235 | 90.0 | 90.0 | 90.0 |
| L1_0--AuCu | dynamic | -2.3279 | -2.3299 | 4.6132 | 4.6132 | 6.6373 | 90.0 | 90.0 | 90.0 |
| B2--CsCl | box | -2.1289 | -2.1309 | 5.2008 | 5.2008 | 5.2008 | 90.0 | 90.0 | 90.0 |
| B1--NaCl--rock-salt | static | -2.0748 | -2.0768 | 5.8144 | 5.8144 | 5.8144 | 90.0 | 90.0 | 90.0 |
| B1--NaCl--rock-salt | static | -1.4375 | -1.4395 | 7.04 | 7.04 | 7.04 | 90.0 | 90.0 | 90.0 |
| B1--NaCl--rock-salt | dynamic | -1.4346 | -1.4366 | 9.1432 | 9.1432 | 9.1432 | 90.0 | 90.0 | 90.0 |
| B1--NaCl--rock-salt | box | -1.3631 | -1.3651 | 7.3198 | 7.3198 | 7.3198 | 90.0 | 90.0 | 90.0 |
| B3--ZnS--cubic-zinc-blende | dynamic | -0.9838 | -0.9858 | 10.9349 | 10.9349 | 10.9349 | 90.0 | 90.0 | 90.0 |
| B3--ZnS--cubic-zinc-blende | static | -0.9035 | -0.9055 | 7.2053 | 7.2053 | 7.2053 | 90.0 | 90.0 | 90.0 |
| B3--ZnS--cubic-zinc-blende | box | -0.5209 | -0.5229 | 8.0459 | 8.0459 | 8.0459 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-1102640 | dynamic | -4.9837 | -4.9864 | 6.3446 | 2.8614 | 9.1462 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | static | -4.2325 | -4.2351 | 5.9759 | 5.9759 | 5.9759 | 90.0 | 90.0 | 90.0 |
| mp-1102640 | box | -4.0851 | -4.0878 | 5.3478 | 3.9727 | 8.1039 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | static | -1.3551 | -1.3578 | 10.7162 | 10.7162 | 10.7162 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | static | -0.5523 | -0.555 | 7.5748 | 7.5748 | 7.5748 | 90.0 | 90.0 | 90.0 |
| C1--CaF2--fluorite | box | -0.519 | -0.5217 | 7.8215 | 7.8215 | 7.8215 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
D0_3--BiF3 = oqmd-309239
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| mp-1221576 | static | -4.6898 | -4.6929 | 2.6857 | 2.6857 | 27.5607 | 90.0 | 90.0 | 120.0 |
| mp-1221576 | box | -4.6676 | -4.6706 | 2.6947 | 2.6947 | 27.4096 | 90.0 | 90.0 | 120.0 |
| mp-31138 | dynamic | -4.2994 | -4.3025 | 3.889 | 3.889 | 15.6313 | 90.0 | 90.0 | 90.0 |
| oqmd-17678 | dynamic | -4.2994 | -4.3025 | 3.889 | 3.889 | 15.6311 | 90.0 | 90.0 | 90.0 |
| mp-31138 | box | -4.298 | -4.301 | 3.8865 | 3.8865 | 15.6551 | 90.0 | 90.0 | 90.0 |
| oqmd-17678 | box | -4.2968 | -4.2998 | 3.8861 | 3.8861 | 15.6623 | 90.0 | 90.0 | 90.0 |
| oqmd-298701 | dynamic | -4.2948 | -4.2978 | 3.8809 | 3.8809 | 7.8691 | 90.0 | 90.0 | 90.0 |
| L1_2--AuCu3 | dynamic | -4.2932 | -4.2963 | 3.8901 | 3.8901 | 3.8901 | 90.0 | 90.0 | 90.0 |
| mp-1186013 | dynamic | -4.2425 | -4.2455 | 5.5219 | 5.5219 | 4.3334 | 90.0 | 90.0 | 120.0 |
| mp-1186013 | box | -4.2361 | -4.2392 | 5.5141 | 5.5141 | 4.3429 | 90.0 | 90.0 | 120.0 |
| D0_3--BiF3 | dynamic | -4.1967 | -4.1998 | 6.032 | 6.032 | 6.032 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | static | -4.0228 | -4.0258 | 4.8936 | 4.8936 | 4.8936 | 90.0 | 90.0 | 90.0 |
| L1_2--AuCu3 | dynamic | -2.3856 | -2.3887 | 6.3942 | 6.3942 | 6.3942 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | static | -2.1249 | -2.128 | 10.0529 | 10.0529 | 10.0529 | 90.0 | 90.0 | 90.0 |
| A15--Cr3Si | static | -2.0313 | -2.0344 | 8.0471 | 8.0471 | 8.0471 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | static | -0.6002 | -0.6033 | 7.7596 | 7.7596 | 7.7596 | 90.0 | 90.0 | 90.0 |
| L1_2--AuCu3 | static | -0.5869 | -0.5899 | 5.2609 | 5.2609 | 5.2609 | 90.0 | 90.0 | 90.0 |
| D0_3--BiF3 | box | -0.5777 | -0.5807 | 7.9414 | 7.9414 | 7.9414 | 90.0 | 90.0 | 90.0 |
Download raw data (including filtered results)
Reference structure matches:
A1--Cu--fcc = mp-2, oqmd-676153, oqmd-1214557
A15--beta-W = oqmd-1215002, oqmd-1280323
A2--W--bcc = oqmd-690547, oqmd-1215180
A3'--alpha-La--double-hcp = mp-1186427, oqmd-1215448
A3--Mg--hcp = oqmd-1215358
A4--C--dc = oqmd-1215537
A5--beta-Sn = oqmd-1215626
A6--In--bct = oqmd-1215715
| prototype | method | Ecoh (eV/atom) | Epot (eV/atom) | a0 (Å) | b0 (Å) | c0 (Å) | α (degrees) | β (degrees) | γ (degrees) |
|---|---|---|---|---|---|---|---|---|---|
| A1--Cu--fcc | dynamic | -5.1945 | -5.1986 | 3.9211 | 3.9211 | 3.9211 | 90.0 | 90.0 | 90.0 |
| A3--Mg--hcp | dynamic | -5.1372 | -5.1413 | 2.8041 | 2.8041 | 4.1927 | 90.0 | 90.0 | 120.0 |
| oqmd-1216074 | dynamic | -5.135 | -5.1391 | 2.8076 | 2.8076 | 19.0612 | 90.0 | 90.0 | 120.0 |
| oqmd-1216074 | static | -5.1341 | -5.1382 | 2.8071 | 2.8071 | 19.0574 | 90.0 | 90.0 | 120.0 |
| A3'--alpha-La--double-hcp | dynamic | -5.1305 | -5.1346 | 2.8036 | 2.8036 | 8.5514 | 90.0 | 90.0 | 120.0 |
| oqmd-1215269 | box | -5.1079 | -5.112 | 2.7902 | 4.8472 | 4.2103 | 90.0 | 90.0 | 90.0 |
| oqmd-1214913 | dynamic | -4.9791 | -4.9832 | 6.76 | 6.76 | 6.76 | 90.0 | 90.0 | 90.0 |
| A2--W--bcc | static | -4.9756 | -4.9797 | 2.9757 | 2.9757 | 2.9757 | 90.0 | 90.0 | 90.0 |
| oqmd-1214913 | box | -4.9611 | -4.9652 | 6.5888 | 6.5888 | 6.5888 | 90.0 | 90.0 | 90.0 |
| oqmd-1214735 | box | -4.928 | -4.9321 | 3.9402 | 8.8745 | 3.9402 | 90.0 | 90.0 | 90.0 |
| A15--beta-W | static | -4.9194 | -4.9236 | 4.8458 | 4.8458 | 4.8458 | 90.0 | 90.0 | 90.0 |
| oqmd-1214824 | dynamic | -4.9012 | -4.9053 | 9.4951 | 9.4951 | 9.4951 | 90.0 | 90.0 | 90.0 |
| oqmd-1214824 | box | -4.8993 | -4.9034 | 9.5038 | 9.5038 | 9.5038 | 90.0 | 90.0 | 90.0 |
| oqmd-1215091 | box | -4.5065 | -4.5106 | 4.078 | 9.3828 | 3.2125 | 90.0 | 90.0 | 90.0 |
| Ah--alpha-Po--sc | static | -4.2733 | -4.2775 | 2.6761 | 2.6761 | 2.6761 | 90.0 | 90.0 | 90.0 |
| Ah--alpha-Po--sc | dynamic | -4.2717 | -4.2758 | 2.7952 | 2.7952 | 2.7952 | 90.0 | 90.0 | 90.0 |
| A5--beta-Sn | static | -4.1851 | -4.1892 | 5.524 | 5.524 | 2.5636 | 90.0 | 90.0 | 90.0 |
| A3--Mg--hcp | box | -4.1376 | -4.1417 | 3.3402 | 3.3402 | 3.2834 | 90.0 | 90.0 | 120.0 |
| A4--C--dc | dynamic | -4.1315 | -4.1356 | 5.926 | 5.926 | 5.926 | 90.0 | 90.0 | 90.0 |
| A7--alpha-As | box | -3.9588 | -3.9629 | 3.9826 | 3.9826 | 10.6294 | 90.0 | 90.0 | 120.0 |
| A5--beta-Sn | box | -2.9165 | -2.9206 | 8.4523 | 8.4523 | 2.5226 | 90.0 | 90.0 | 90.0 |
| A1--Cu--fcc | dynamic | -2.6209 | -2.6251 | 6.208 | 6.208 | 6.208 | 90.0 | 90.0 | 90.0 |
| A3'--alpha-La--double-hcp | dynamic | -2.6209 | -2.6251 | 4.3897 | 4.3897 | 14.3368 | 90.0 | 90.0 | 120.0 |
| A3--Mg--hcp | dynamic | -2.6209 | -2.6251 | 4.3897 | 4.3897 | 7.1684 | 90.0 | 90.0 | 120.0 |
| A2--W--bcc | dynamic | -2.5842 | -2.5884 | 4.5343 | 4.5343 | 4.5343 | 90.0 | 90.0 | 90.0 |
| A15--beta-W | static | -2.5592 | -2.5633 | 6.9885 | 6.9885 | 6.9885 | 90.0 | 90.0 | 90.0 |
| A1--Cu--fcc | static | -2.0453 | -2.0495 | 5.1569 | 5.1569 | 5.1569 | 90.0 | 90.0 | 90.0 |
| A3'--alpha-La--double-hcp | box | -1.9745 | -1.9787 | 3.6359 | 3.6359 | 12.2073 | 90.0 | 90.0 | 120.0 |
| A5--beta-Sn | static | -1.9368 | -1.9409 | 8.2405 | 8.2405 | 3.926 | 90.0 | 90.0 | 90.0 |
| Ah--alpha-Po--sc | dynamic | -1.8794 | -1.8835 | 4.4142 | 4.4142 | 4.4142 | 90.0 | 90.0 | 90.0 |
| A3--Mg--hcp | box | -1.4969 | -1.501 | 4.3573 | 4.3573 | 9.9779 | 90.0 | 90.0 | 120.0 |
| oqmd-1215982 | box | -1.388 | -1.3921 | 3.6848 | 3.6848 | 4.8766 | 90.0 | 90.0 | 120.0 |
| A4--C--dc | dynamic | -1.2172 | -1.2213 | 9.0426 | 9.0426 | 9.0426 | 90.0 | 90.0 | 90.0 |
| A4--C--dc | dynamic | -1.0789 | -1.083 | 10.0586 | 10.0586 | 10.0586 | 90.0 | 90.0 | 90.0 |
| A5--beta-Sn | box | -0.6375 | -0.6416 | 5.8901 | 5.8901 | 7.2183 | 90.0 | 90.0 | 90.0 |
| A15--beta-W | static | -0.5736 | -0.5777 | 10.3057 | 10.3057 | 10.3057 | 90.0 | 90.0 | 90.0 |
| A3'--alpha-La--double-hcp | box | 3.3559 | 3.3518 | 3.1953 | 3.1953 | 4.2466 | 90.0 | 90.0 | 120.0 |
Elastic Constants Predictions
Static elastic constants are displayed for the unique structures identified in Crystal Structure Predictions above. The values displayed here are obtained by measuring the change in virial stresses due to applying small strains to the relaxed crystals. The initial structure and the strained states are all relaxed using force minimization.
The calculation method used is available as the iprPy elastic_constants_static calculation method.
Notes and Disclaimers:
- These values are meant to be guidelines for comparing potentials, not the absolute values for any potential's properties. Values listed here may change if the calculation methods are updated due to improvements/corrections. Variations in the values may occur for variations in calculation methods, simulation software and implementations of the interatomic potentials.
- The presence of any structures in this list does not guarantee that those structures are stable.
- The elastic constants have been computed for a variety of strains, and in some cases for slightly different lattice constant values. The static nature of this calculation can give poor predictions if the evaluated states straddle a functional discontinuity in the potential's third derivative. Be sure to compare the elastic constants for the different strains (positive and negative).
- NIST disclaimer
Version Information:
- 2019-08-07. Data added.
| 55.698 | 29.874 | 29.874 | 0.000 | 0.000 | 0.000 |
| 29.874 | 55.698 | 29.874 | 0.000 | 0.000 | 0.000 |
| 29.874 | 29.874 | 55.698 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 8.982 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 8.982 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 8.982 |
| 55.698 | 29.874 | 29.874 | -0.000 | -0.000 | -0.000 |
| 29.874 | 55.698 | 29.874 | -0.000 | -0.000 | -0.000 |
| 29.874 | 29.874 | 55.698 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 8.982 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 8.982 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 8.982 |
| 55.698 | 29.874 | 29.874 | 0.000 | 0.000 | -0.000 |
| 29.874 | 55.698 | 29.874 | 0.000 | -0.000 | 0.000 |
| 29.874 | 29.874 | 55.698 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 8.982 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 8.982 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 8.982 |
| 55.698 | 29.874 | 29.874 | -0.000 | 0.000 | -0.000 |
| 29.874 | 55.698 | 29.874 | -0.000 | 0.000 | -0.000 |
| 29.874 | 29.874 | 55.698 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 8.982 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 8.982 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 8.982 |
| 55.697 | 29.873 | 29.873 | 0.000 | 0.000 | 0.000 |
| 29.873 | 55.697 | 29.873 | 0.000 | 0.000 | 0.000 |
| 29.873 | 29.873 | 55.697 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 8.982 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 8.982 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 8.982 |
| 55.698 | 29.874 | 29.874 | -0.000 | -0.000 | -0.000 |
| 29.874 | 55.698 | 29.874 | -0.000 | -0.000 | -0.000 |
| 29.874 | 29.874 | 55.698 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 8.982 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 8.982 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 8.982 |
| 46.040 | 9.914 | 9.914 | -0.000 | -0.000 | 0.000 |
| 9.914 | 46.040 | 9.914 | -0.000 | 0.000 | -0.000 |
| 9.914 | 9.914 | 46.040 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -17.651 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -17.651 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -17.651 |
| 46.040 | 9.914 | 9.914 | 0.000 | -0.000 | -0.000 |
| 9.914 | 46.040 | 9.914 | 0.000 | 0.000 | -0.000 |
| 9.914 | 9.914 | 46.040 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -17.651 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -17.651 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -0.000 | -17.651 |
| 46.040 | 9.914 | 9.914 | -0.000 | -0.000 | -0.000 |
| 9.914 | 46.040 | 9.914 | -0.000 | -0.000 | -0.000 |
| 9.914 | 9.914 | 46.040 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -12.163 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -12.163 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -12.163 |
| 46.040 | 9.914 | 9.914 | 0.000 | -0.000 | -0.000 |
| 9.914 | 46.040 | 9.914 | 0.000 | -0.000 | -0.000 |
| 9.914 | 9.914 | 46.040 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -12.163 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -12.163 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -12.163 |
| 45.502 | 10.184 | 10.184 | -0.000 | 0.000 | 0.000 |
| 10.184 | 45.502 | 10.184 | -0.000 | -0.000 | 0.000 |
| 10.184 | 10.184 | 45.502 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -17.651 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -17.651 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -17.651 |
| 46.039 | 9.914 | 9.914 | 0.000 | 0.000 | -0.000 |
| 9.914 | 46.039 | 9.914 | 0.000 | 0.000 | 0.000 |
| 9.914 | 9.914 | 46.039 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -17.651 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -17.651 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -17.651 |
| 63.106 | 57.155 | 57.155 | -0.000 | 0.000 | -0.000 |
| 57.155 | 63.106 | 57.155 | -0.000 | 0.000 | -0.000 |
| 57.155 | 57.155 | 63.106 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 33.106 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 33.106 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 33.106 |
| 63.106 | 57.155 | 57.155 | 0.000 | 0.000 | 0.000 |
| 57.155 | 63.106 | 57.155 | 0.000 | 0.000 | 0.000 |
| 57.155 | 57.155 | 63.106 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 33.106 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 33.106 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 33.106 |
| 63.106 | 57.155 | 57.155 | 0.000 | 0.000 | -0.000 |
| 57.155 | 63.106 | 57.155 | 0.000 | -0.000 | 0.000 |
| 57.155 | 57.155 | 63.106 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 33.106 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 33.106 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 33.106 |
| 63.106 | 57.155 | 57.155 | 0.000 | -0.000 | 0.000 |
| 57.155 | 63.106 | 57.155 | -0.000 | 0.000 | 0.000 |
| 57.155 | 57.155 | 63.106 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 33.106 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 33.106 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 33.106 |
| 63.106 | 57.155 | 57.155 | -0.000 | -0.000 | 0.000 |
| 57.155 | 63.106 | 57.155 | 0.000 | -0.000 | -0.000 |
| 57.155 | 57.155 | 63.106 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 33.106 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 33.106 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 33.106 |
| 63.106 | 57.155 | 57.155 | 0.000 | -0.000 | 0.000 |
| 57.155 | 63.106 | 57.155 | -0.000 | -0.000 | 0.000 |
| 57.155 | 57.155 | 63.106 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 33.106 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 33.106 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 33.106 |
| 50.190 | 30.318 | 40.988 | -0.000 | 0.000 | 0.000 |
| 30.256 | 50.252 | 40.988 | 0.000 | 0.000 | 0.000 |
| 40.988 | 40.988 | 74.275 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 20.421 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 20.421 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 9.936 |
| 50.252 | 30.256 | 40.988 | 0.000 | 0.000 | -0.000 |
| 30.318 | 50.190 | 40.988 | 0.000 | 0.000 | 0.000 |
| 40.988 | 40.988 | 74.275 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 20.421 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 20.421 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 9.936 |
| 50.190 | 30.318 | 40.988 | -0.000 | -0.000 | 0.000 |
| 30.318 | 50.190 | 40.988 | -0.000 | -0.000 | 0.000 |
| 40.988 | 40.988 | 74.275 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 20.421 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 20.421 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 9.936 |
| 50.252 | 30.256 | 40.988 | 0.000 | -0.000 | -0.000 |
| 30.256 | 50.252 | 40.988 | 0.000 | 0.000 | -0.000 |
| 40.988 | 40.988 | 74.275 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 20.421 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 20.421 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 9.936 |
| 50.172 | 30.336 | 40.988 | -0.000 | -0.000 | 0.000 |
| 30.318 | 50.190 | 40.988 | -0.000 | -0.000 | 0.000 |
| 40.988 | 40.988 | 74.276 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 20.421 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 20.421 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 9.998 |
| 50.251 | 30.256 | 40.988 | 0.000 | 0.000 | 0.000 |
| 30.336 | 50.171 | 40.988 | 0.000 | 0.000 | -0.000 |
| 40.988 | 40.988 | 74.274 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 20.421 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 20.421 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 9.998 |
| 49.817 | 29.714 | 46.100 | 0.000 | 0.000 | -0.000 |
| 29.309 | 50.222 | 46.100 | 0.000 | 0.000 | 0.000 |
| 46.100 | 46.100 | 102.490 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 27.018 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 27.018 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 10.457 |
| 50.222 | 29.309 | 46.100 | 0.000 | 0.000 | -0.000 |
| 29.309 | 50.222 | 46.100 | -0.000 | -0.000 | -0.000 |
| 46.100 | 46.100 | 102.490 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 27.018 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 27.018 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 10.457 |
| 50.222 | 29.308 | 46.100 | -0.000 | -0.000 | 0.000 |
| 29.308 | 50.222 | 46.100 | 0.000 | 0.000 | 0.000 |
| 46.100 | 46.100 | 102.490 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 27.018 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 27.018 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 10.052 |
| 49.817 | 29.714 | 46.100 | -0.000 | -0.000 | 0.000 |
| 29.714 | 49.817 | 46.100 | -0.000 | -0.000 | 0.000 |
| 46.100 | 46.100 | 102.490 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 27.018 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 27.018 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 10.052 |
| 49.817 | 29.713 | 46.100 | 0.000 | 0.000 | -0.000 |
| 29.713 | 49.816 | 46.100 | 0.000 | 0.000 | -0.000 |
| 46.099 | 46.099 | 102.490 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 27.018 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 27.018 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 10.457 |
| 49.818 | 29.714 | 46.101 | -0.000 | -0.000 | 0.000 |
| 29.309 | 50.223 | 46.101 | 0.000 | 0.000 | -0.000 |
| 46.100 | 46.100 | 102.490 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 27.018 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 27.018 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 10.457 |
| 50.296 | 29.381 | 46.171 | 0.000 | -0.000 | 0.000 |
| 29.787 | 49.890 | 46.171 | 0.000 | 0.000 | -0.000 |
| 46.171 | 46.171 | 102.544 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 27.016 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 27.016 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 10.052 |
| 50.296 | 29.381 | 46.171 | -0.000 | 0.000 | 0.000 |
| 29.381 | 50.296 | 46.171 | -0.000 | -0.000 | 0.000 |
| 46.171 | 46.171 | 102.544 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 27.016 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 27.016 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 10.052 |
| 49.890 | 29.787 | 46.170 | 0.000 | 0.000 | -0.000 |
| 29.381 | 50.296 | 46.170 | 0.000 | 0.000 | 0.000 |
| 46.170 | 46.170 | 102.544 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 27.016 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 27.016 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 10.457 |
| 50.296 | 29.381 | 46.171 | -0.000 | -0.000 | -0.000 |
| 29.381 | 50.296 | 46.171 | -0.000 | 0.000 | 0.000 |
| 46.171 | 46.171 | 102.544 | -0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 27.016 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 27.016 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 10.457 |
| 49.890 | 29.786 | 46.170 | 0.000 | -0.000 | -0.000 |
| 29.786 | 49.890 | 46.170 | 0.000 | -0.000 | -0.000 |
| 46.170 | 46.170 | 102.544 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 27.016 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 27.016 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 10.457 |
| 50.297 | 29.382 | 46.171 | -0.000 | 0.000 | 0.000 |
| 29.382 | 50.297 | 46.171 | -0.000 | -0.000 | 0.000 |
| 46.171 | 46.171 | 102.544 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 27.016 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 27.016 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 10.457 |
| 20.363 | 36.162 | 36.162 | 0.000 | 0.000 | 0.000 |
| 36.162 | 20.363 | 36.162 | 0.000 | 0.000 | 0.000 |
| 36.162 | 36.162 | 20.363 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 27.109 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 27.109 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 27.109 |
| 20.363 | 36.162 | 36.162 | -0.000 | -0.000 | -0.000 |
| 36.162 | 20.363 | 36.162 | -0.000 | -0.000 | -0.000 |
| 36.162 | 36.162 | 20.363 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 27.109 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 27.109 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 27.109 |
| 20.363 | 36.162 | 36.162 | 0.000 | 0.000 | 0.000 |
| 36.162 | 20.363 | 36.162 | 0.000 | 0.000 | 0.000 |
| 36.162 | 36.162 | 20.363 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -12.253 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -12.253 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -12.253 |
| 20.363 | 36.162 | 36.162 | -0.000 | -0.000 | -0.000 |
| 36.162 | 20.363 | 36.162 | -0.000 | -0.000 | -0.000 |
| 36.162 | 36.162 | 20.363 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -12.253 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -12.253 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -12.253 |
| 20.363 | 36.162 | 36.162 | 0.000 | 0.000 | 0.000 |
| 36.162 | 20.363 | 36.162 | 0.000 | 0.000 | 0.000 |
| 36.162 | 36.162 | 20.363 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -12.253 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -12.253 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -12.253 |
| 20.363 | 36.162 | 36.162 | -0.000 | -0.000 | -0.000 |
| 36.162 | 20.363 | 36.162 | -0.000 | -0.000 | -0.000 |
| 36.162 | 36.162 | 20.363 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -12.253 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -12.253 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -12.253 |
| 71.378 | 16.542 | 15.350 | 0.000 | -0.000 | 0.000 |
| 16.542 | 71.378 | 15.350 | 0.000 | -0.000 | 0.000 |
| 15.350 | 15.350 | 112.817 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -3.292 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -3.292 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 6.516 |
| 71.378 | 16.542 | 15.350 | -0.000 | -0.000 | 0.000 |
| 16.542 | 71.378 | 15.350 | 0.000 | -0.000 | -0.000 |
| 15.350 | 15.350 | 112.817 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -3.292 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -3.292 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 6.516 |
| 20.711 | 67.209 | 15.350 | -0.000 | 0.000 | 0.000 |
| 67.209 | 20.711 | 15.350 | 0.000 | -0.000 | -0.000 |
| 15.350 | 15.350 | 112.817 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -3.292 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -3.292 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 6.516 |
| 20.711 | 67.209 | 15.350 | 0.000 | 0.000 | 0.000 |
| 16.542 | 71.378 | 15.350 | -0.000 | 0.000 | -0.000 |
| 15.350 | 15.350 | 112.816 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -3.292 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -3.292 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 6.516 |
| 71.378 | 16.542 | 15.350 | -0.000 | -0.000 | 0.000 |
| 16.542 | 71.378 | 15.350 | -0.000 | -0.000 | 0.000 |
| 15.350 | 15.350 | 112.818 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -3.292 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -3.292 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 6.516 |
| 43.893 | 44.027 | 15.350 | -0.000 | 0.000 | 0.000 |
| 44.027 | 43.893 | 15.350 | 0.000 | 0.000 | 0.000 |
| 15.350 | 15.350 | 112.815 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -3.292 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -3.292 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 6.516 |
| 77.450 | 9.391 | 9.391 | 0.000 | -0.000 | 0.000 |
| 9.391 | 77.450 | 9.391 | 0.000 | 0.000 | 0.000 |
| 9.391 | 9.391 | 77.450 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.110 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.110 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 0.110 |
| 77.450 | 9.391 | 9.391 | -0.000 | -0.000 | -0.000 |
| 9.391 | 77.450 | 9.391 | 0.000 | 0.000 | -0.000 |
| 9.391 | 9.391 | 77.450 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.110 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.110 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 0.110 |
| 77.450 | 9.391 | 9.391 | 0.000 | -0.000 | 0.000 |
| 9.391 | 77.450 | 9.391 | 0.000 | 0.000 | 0.000 |
| 9.391 | 9.391 | 77.450 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.110 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.110 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 0.110 |
| 77.450 | 9.391 | 9.391 | -0.000 | 0.000 | -0.000 |
| 9.391 | 77.450 | 9.391 | -0.000 | -0.000 | -0.000 |
| 9.391 | 9.391 | 77.450 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.110 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.110 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 0.110 |
| 77.450 | 9.390 | 9.390 | 0.000 | 0.000 | 0.000 |
| 9.390 | 77.450 | 9.390 | 0.000 | -0.000 | 0.000 |
| 9.390 | 9.390 | 77.450 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.110 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.110 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 0.110 |
| 77.449 | 9.391 | 9.391 | -0.000 | -0.000 | -0.000 |
| 9.391 | 77.449 | 9.391 | -0.000 | -0.000 | -0.000 |
| 9.391 | 9.391 | 77.449 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.110 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.110 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 0.110 |
| 29.162 | 25.655 | 15.926 | -0.000 | 0.000 | -0.000 |
| 25.655 | 29.162 | 15.926 | -0.000 | 0.000 | -0.000 |
| 16.751 | 16.751 | 52.414 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 3.605 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 3.605 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 15.412 |
| 8.290 | 41.789 | 17.154 | -0.000 | -0.000 | -0.000 |
| 41.789 | 8.290 | 17.154 | -0.000 | -0.000 | 0.000 |
| 15.523 | 15.523 | 53.051 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 3.605 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 3.605 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 15.412 |
| 29.162 | 25.655 | 15.926 | -0.000 | -0.000 | 0.000 |
| 25.655 | 29.162 | 15.926 | -0.000 | 0.000 | 0.000 |
| 15.523 | 15.523 | 53.051 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.166 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.166 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 15.412 |
| 20.835 | 25.607 | 18.097 | 0.000 | 0.000 | 0.000 |
| 25.607 | 20.835 | 18.097 | -0.000 | 0.000 | -0.000 |
| 16.751 | 16.751 | 52.414 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.166 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.166 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 15.412 |
| 29.162 | 25.655 | 15.926 | -0.000 | 0.000 | -0.000 |
| 25.655 | 29.162 | 15.926 | -0.000 | 0.000 | -0.000 |
| 15.523 | 15.523 | 53.051 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.166 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 0.166 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 15.412 |
| 8.290 | 41.789 | 17.154 | -0.000 | -0.000 | -0.000 |
| 41.789 | 8.290 | 17.154 | -0.000 | -0.000 | -0.000 |
| 16.751 | 16.751 | 52.414 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.166 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.166 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 15.412 |
| 66.797 | 33.411 | 33.722 | -0.638 | -2.489 | -2.802 |
| 180.555 | 125.981 | -103.682 | 23.398 | -271.983 | 33.140 |
| 13.046 | 41.246 | 57.684 | 13.726 | -142.447 | 21.507 |
| -0.706 | -0.483 | 1.194 | 15.519 | -0.111 | -0.384 |
| -20.733 | 6.744 | -8.479 | 12.394 | -129.791 | 21.308 |
| -1.134 | -1.037 | -0.329 | -0.384 | -1.473 | 15.243 |
| -182.960 | 13.457 | 226.284 | 32.427 | 273.514 | 3.521 |
| -111.280 | -18.405 | 174.776 | -24.292 | 272.293 | -35.939 |
| -200.816 | 14.763 | 248.316 | 35.051 | 275.011 | 5.571 |
| 20.585 | -7.055 | 10.640 | 0.637 | 143.248 | -22.158 |
| 0.698 | -0.308 | 1.001 | -0.111 | 15.756 | -0.814 |
| -1.134 | -1.037 | -0.329 | -0.384 | -1.473 | 15.243 |
| 66.797 | 33.411 | 33.722 | -0.638 | -2.489 | -2.802 |
| 33.236 | 65.845 | 33.674 | -1.138 | -0.308 | -1.034 |
| 33.885 | 33.726 | 70.776 | 1.194 | 1.001 | -0.286 |
| 13.827 | 7.117 | -12.066 | 11.331 | -27.164 | 3.160 |
| 0.698 | -0.308 | 1.001 | -0.111 | 15.756 | -0.814 |
| 13.164 | 5.776 | -14.052 | 2.087 | -27.906 | 12.722 |
| 29.370 | 32.523 | 54.092 | 2.604 | 26.990 | -1.643 |
| 20.048 | 46.587 | 49.462 | -2.832 | 27.359 | -4.852 |
| 33.885 | 33.726 | 70.776 | 1.194 | 1.001 | -0.286 |
| -0.706 | -0.483 | 1.194 | 15.519 | -0.111 | -0.384 |
| -22.619 | -1.904 | 20.216 | 3.413 | 34.523 | -0.066 |
| -1.134 | -1.037 | -0.329 | -0.384 | -1.473 | 15.243 |
| 55.904 | 34.765 | 33.132 | -1.026 | -3.102 | -2.309 |
| 33.236 | 65.845 | 33.674 | -1.138 | -0.308 | -1.034 |
| 33.886 | 33.726 | 70.776 | 1.194 | 1.001 | -0.286 |
| -1.086 | -0.190 | 1.188 | 13.172 | -1.500 | -0.209 |
| 0.501 | 0.024 | 0.783 | 0.078 | 12.021 | -0.196 |
| -1.410 | -1.100 | -0.358 | -0.306 | -2.429 | 13.053 |
| 63.303 | 34.080 | 34.492 | -0.948 | -0.507 | -2.873 |
| 34.236 | 61.654 | 34.523 | -0.931 | 1.454 | -1.383 |
| 33.885 | 33.726 | 70.775 | 1.194 | 1.001 | -0.286 |
| -0.706 | -0.483 | 1.194 | 15.519 | -0.111 | -0.384 |
| 0.698 | -0.308 | 1.001 | -0.111 | 15.756 | -0.814 |
| -0.977 | -1.237 | -0.169 | -0.557 | 0.463 | 12.623 |
| 70.479 | 34.066 | 33.847 | -0.249 | -2.630 | -1.769 |
| 34.274 | 66.635 | 33.430 | -2.059 | 0.089 | 0.658 |
| 228.324 | 595.521 | 100.021 | 586.782 | 808.939 | 702.119 |
| 206.756 | -121.248 | 57.127 | -7.177 | 74.690 | 27.930 |
| 206.039 | -80.774 | 47.359 | 20.301 | 111.180 | 81.241 |
| 206.023 | -119.394 | 56.738 | -20.301 | 73.742 | 41.914 |
| 70.479 | 34.066 | 33.847 | -0.249 | -2.630 | -1.769 |
| -293.582 | -248.400 | -37.662 | -405.434 | -634.602 | -639.200 |
| -78.396 | -44.325 | 42.900 | -171.653 | -217.988 | -167.377 |
| -115.443 | -80.760 | -9.700 | -163.049 | -217.547 | -167.045 |
| -0.304 | -0.048 | 0.533 | 0.044 | 15.863 | -0.222 |
| -1.025 | 0.636 | 0.149 | -0.223 | -1.338 | 16.230 |
| 70.479 | 34.066 | 33.847 | -0.249 | -2.630 | -1.769 |
| 56.039 | 51.535 | 39.507 | -2.200 | 9.145 | 6.007 |
| 33.944 | 33.271 | 67.868 | 0.138 | 0.533 | 0.223 |
| 20.314 | -13.201 | 6.156 | 10.993 | 7.461 | 2.556 |
| 20.222 | -11.817 | 6.057 | -2.016 | 20.423 | 2.784 |
| -1.025 | 0.636 | 0.149 | -0.223 | -1.338 | 16.230 |
| 70.479 | 34.066 | 33.847 | -0.249 | -2.630 | -1.769 |
| 34.274 | 66.635 | 33.430 | -2.059 | 0.089 | 0.658 |
| 14.238 | 46.090 | 57.885 | 2.519 | -7.072 | -2.641 |
| -20.840 | -6.060 | -1.466 | -7.465 | -18.652 | -25.431 |
| -0.304 | -0.048 | 0.533 | 0.044 | 15.863 | -0.222 |
| -1.025 | 0.636 | 0.149 | -0.223 | -1.338 | 16.230 |
| 61.984 | 35.115 | 36.558 | 0.245 | -0.744 | -0.251 |
| 37.340 | 61.442 | 34.796 | -2.409 | 1.076 | 0.952 |
| 37.145 | 32.874 | 63.953 | 0.307 | 1.167 | 0.469 |
| 1.698 | -2.413 | 1.066 | 12.808 | 0.748 | 0.023 |
| 1.618 | -1.034 | 0.970 | -0.204 | 13.659 | 0.257 |
| -1.025 | 0.637 | 0.149 | -0.223 | -1.338 | 16.230 |
| 70.479 | 34.066 | 33.847 | -0.249 | -2.630 | -1.769 |
| 33.126 | 63.720 | 33.669 | -2.001 | -0.449 | 0.372 |
| 32.847 | 35.341 | 62.857 | 0.717 | -0.374 | -0.113 |
| -0.216 | -0.902 | 0.138 | 15.385 | 0.044 | -0.359 |
| -2.649 | 1.371 | -0.203 | 0.103 | 11.602 | -0.422 |
| -3.615 | -2.159 | -0.300 | -2.972 | -3.641 | 3.478 |
| -0.944 | 280.372 | 86.330 | -328.393 | -100.183 | 220.501 |
| 34.814 | 64.909 | 33.125 | -1.592 | 0.708 | 0.604 |
| 31.983 | 33.188 | 63.118 | -1.141 | 1.774 | 1.467 |
| -0.489 | -2.496 | -1.141 | 16.143 | 1.486 | 0.701 |
| -55.626 | 244.397 | 53.075 | -327.101 | -93.946 | 218.456 |
| -0.602 | 0.600 | 1.569 | 0.781 | 0.358 | 17.783 |
| 131.327 | -67.792 | 83.003 | 175.606 | -44.930 | -184.775 |
| 34.814 | 64.909 | 33.125 | -1.592 | 0.708 | 0.604 |
| 31.983 | 33.188 | 63.118 | -1.141 | 1.774 | 1.467 |
| 54.443 | -246.847 | -53.709 | 337.042 | 101.409 | -218.975 |
| -1.345 | 0.628 | 1.774 | 1.486 | 14.841 | -0.468 |
| -0.602 | 0.600 | 1.569 | 0.781 | 0.358 | 17.783 |
| 48.396 | 47.447 | 29.790 | -17.741 | 4.592 | 19.598 |
| 43.203 | 56.178 | 27.584 | 1.938 | 0.140 | 3.145 |
| 26.871 | 45.968 | 47.864 | -19.018 | 5.757 | 19.151 |
| -7.939 | 7.187 | -6.329 | -7.464 | 6.022 | 19.242 |
| -8.501 | 10.744 | -3.582 | -16.103 | 11.886 | 18.037 |
| -25.604 | 39.630 | 26.017 | -60.663 | -29.704 | 41.091 |
| 63.498 | 26.493 | 39.459 | 17.411 | -4.410 | -17.560 |
| 28.895 | 62.007 | 40.911 | -6.094 | 0.664 | -1.528 |
| 39.830 | 11.969 | 43.349 | 30.949 | 11.277 | -21.672 |
| 7.136 | -13.761 | 3.348 | 27.708 | -2.982 | -17.913 |
| -8.303 | 3.248 | 8.237 | -2.533 | 11.565 | -2.748 |
| -7.471 | 3.713 | 7.884 | -3.078 | 0.481 | 12.187 |
| 56.784 | 43.788 | 55.797 | -23.479 | -11.718 | 17.640 |
| 34.814 | 64.909 | 33.125 | -1.592 | 0.708 | 0.604 |
| 31.449 | 41.029 | 47.902 | -8.013 | -1.277 | 3.055 |
| 0.197 | -3.129 | -1.880 | 13.593 | 1.457 | 1.111 |
| -1.708 | 1.303 | 0.777 | -0.266 | 7.795 | 1.295 |
| -0.602 | 0.600 | 1.569 | 0.781 | 0.358 | 17.783 |
| 67.119 | 34.974 | 32.346 | -0.223 | 0.769 | 0.633 |
| 37.055 | 47.709 | 35.709 | 0.405 | 1.777 | -1.308 |
| 32.622 | 34.623 | 59.198 | -1.600 | 1.594 | 0.875 |
| -1.237 | -2.522 | -0.539 | 12.963 | 1.510 | 0.632 |
| 1.162 | -2.952 | -1.027 | 7.491 | 7.976 | -3.887 |
| -1.027 | 1.085 | 1.885 | 0.537 | 0.243 | 14.223 |
| -193.367 | 199.784 | 42.577 | 18.824 | 9.921 | -47.379 |
| 33.388 | 64.929 | 33.383 | -2.018 | 0.372 | 0.658 |
| -219.627 | 199.703 | 71.642 | 16.898 | 10.285 | -47.384 |
| -252.675 | 162.413 | 6.928 | 33.133 | 10.428 | -47.681 |
| -313.720 | 218.160 | -119.961 | 31.874 | -8.197 | 14.328 |
| -251.977 | 166.083 | 10.375 | 19.923 | 9.657 | -34.220 |
| 310.940 | -130.705 | 23.994 | -19.687 | -8.389 | 49.231 |
| 33.388 | 64.929 | 33.383 | -2.018 | 0.372 | 0.658 |
| 549.506 | -362.228 | 671.206 | -94.814 | -64.652 | -135.011 |
| 251.633 | -168.077 | -11.655 | -5.379 | -7.882 | 48.928 |
| 251.357 | -165.274 | -8.164 | -17.982 | 2.488 | 48.145 |
| 252.331 | -164.406 | -8.208 | -18.588 | -8.653 | 62.388 |
| 63.246 | 33.840 | 32.209 | -0.336 | 0.899 | 0.926 |
| 33.388 | 64.929 | 33.383 | -2.018 | 0.372 | 0.658 |
| 31.499 | 33.392 | 65.861 | -2.310 | 1.197 | 1.138 |
| -25.740 | 13.695 | -1.434 | 15.802 | 2.188 | -4.207 |
| -26.017 | 16.497 | 2.057 | 3.200 | 12.557 | -4.991 |
| -0.036 | 0.653 | 1.293 | 0.458 | 0.547 | 16.700 |
| 83.999 | 18.013 | 32.358 | -2.358 | -0.150 | 5.758 |
| 59.345 | 43.683 | 33.469 | -4.215 | -0.908 | 5.735 |
| 57.747 | 17.926 | 61.431 | -4.284 | 0.214 | 5.753 |
| 24.698 | -19.358 | -3.293 | 11.951 | 0.357 | 5.455 |
| -1.044 | 0.327 | 1.197 | 1.240 | 14.559 | -0.278 |
| -0.036 | 0.653 | 1.293 | 0.458 | 0.547 | 16.700 |
| 56.197 | 36.229 | 33.384 | -0.235 | 0.859 | 0.433 |
| 33.388 | 64.930 | 33.383 | -2.018 | 0.372 | 0.658 |
| 31.499 | 33.392 | 65.861 | -2.310 | 1.197 | 1.138 |
| -3.437 | 0.234 | -5.953 | 9.656 | 1.184 | 0.835 |
| -3.354 | 1.642 | 1.220 | 1.471 | 11.670 | -0.648 |
| -0.036 | 0.653 | 1.293 | 0.458 | 0.547 | 16.700 |
| 57.159 | 32.729 | 35.519 | -0.677 | 0.803 | 0.851 |
| 36.717 | 58.444 | 34.319 | -2.490 | -0.092 | 1.401 |
| 37.457 | 32.898 | 57.205 | -3.068 | 1.464 | 0.167 |
| 2.030 | -4.506 | -2.458 | 13.654 | 1.181 | 1.115 |
| 1.768 | -1.713 | 1.032 | 1.079 | 11.521 | 0.333 |
| 2.737 | -0.829 | 0.983 | 0.479 | 0.408 | 14.489 |
| 68.761 | 34.677 | 35.072 | 0.006 | 0.804 | 1.904 |
| 42.201 | 80.725 | -117.121 | 86.962 | -68.361 | -33.667 |
| 35.391 | 34.201 | 63.010 | 0.643 | -1.644 | 0.105 |
| 592.027 | -341.908 | -471.294 | 74.766 | 151.973 | 255.356 |
| 3118.473 | -906.756 | -1797.905 | 1066.640 | -1405.056 | -266.448 |
| 7.053 | 13.464 | -151.956 | 87.190 | -68.833 | -19.413 |
| 57.332 | 20.849 | 188.293 | -86.072 | 69.885 | 34.436 |
| 28.485 | 51.974 | 187.161 | -85.051 | 70.232 | 31.885 |
| -2435.810 | 1107.595 | 1476.091 | -335.235 | 829.043 | 136.264 |
| -7.222 | -14.611 | 152.849 | -72.912 | 69.554 | 33.986 |
| -6.791 | -13.426 | 150.784 | -85.756 | 83.739 | 32.256 |
| -2086.008 | 951.024 | 1174.227 | -200.413 | 456.580 | 28.159 |
| 281.250 | -75.947 | -109.664 | 29.750 | -83.946 | -12.615 |
| 36.029 | 67.787 | 19.805 | 9.557 | -5.995 | -4.169 |
| 244.929 | -60.276 | -86.901 | 20.940 | -45.924 | -1.760 |
| -0.299 | -0.034 | 0.643 | 15.803 | 0.192 | 1.247 |
| 0.753 | 2.387 | -16.572 | 8.852 | 7.517 | -3.797 |
| 0.881 | 0.526 | -15.030 | 9.785 | -6.466 | 10.089 |
| -240.862 | 140.354 | 180.097 | -69.327 | 113.609 | 28.273 |
| 34.657 | 64.911 | 50.236 | -7.646 | 7.866 | 2.387 |
| -374.754 | 73.983 | 276.041 | -198.268 | 198.524 | 64.509 |
| -252.714 | 106.918 | 145.576 | -33.111 | 85.600 | 16.280 |
| -0.619 | -0.489 | 13.859 | -8.350 | 21.369 | 2.758 |
| -208.093 | 94.982 | 118.351 | -18.445 | 45.352 | 3.125 |
| 55.052 | 23.097 | 25.063 | -0.602 | -5.201 | -0.024 |
| 40.059 | 50.540 | 31.821 | 0.756 | 2.057 | 2.003 |
| 71.970 | 27.661 | 24.560 | 18.000 | -18.673 | -3.714 |
| 24.854 | -10.039 | -16.491 | 16.076 | -11.451 | 0.987 |
| 0.602 | 1.232 | -2.781 | 0.358 | 12.972 | 1.123 |
| 0.263 | -0.766 | -1.344 | 2.048 | -0.229 | 13.061 |
| 68.760 | 34.677 | 35.072 | 0.006 | 0.804 | 1.904 |
| 34.781 | 70.587 | 34.051 | 1.343 | 1.263 | -0.858 |
| 36.380 | 35.029 | 59.786 | -0.148 | -0.675 | 0.539 |
| -0.433 | -0.378 | 2.235 | 12.245 | 0.953 | 1.539 |
| 0.161 | 1.285 | -1.644 | 0.192 | 16.922 | -0.394 |
| -23.145 | 10.433 | 14.653 | -0.477 | 8.050 | 3.560 |
| 64.231 | 34.379 | 34.279 | 0.981 | 0.106 | 1.162 |
| -3.349 | -0.044 | -15.168 | 1.872 | 16.675 | 92.884 |
| 34.010 | 33.938 | 66.842 | -0.652 | 0.625 | -0.180 |
| 1.020 | -0.007 | -0.652 | 15.856 | -0.145 | -0.107 |
| -0.832 | -0.195 | 0.625 | -0.145 | 16.127 | 0.969 |
| -144.932 | 35.336 | -20.116 | -20.034 | 57.263 | 169.531 |
| 64.231 | 34.379 | 34.279 | 0.981 | 0.106 | 1.162 |
| 34.358 | 64.638 | 34.001 | 1.083 | -0.257 | -0.567 |
| 218.327 | -25.876 | 130.652 | 13.637 | -28.785 | -155.671 |
| 1.020 | -0.007 | -0.652 | 15.856 | -0.145 | -0.107 |
| 27.948 | 67.087 | 48.388 | -1.830 | -2.706 | -88.596 |
| 105.354 | 2.921 | 61.920 | 5.470 | -20.871 | -111.638 |
| 34.096 | 37.775 | 35.240 | -1.442 | 5.009 | 16.322 |
| 34.358 | 64.638 | 34.001 | 1.083 | -0.257 | -0.567 |
| 32.394 | 27.663 | 57.877 | -0.313 | 2.127 | 8.975 |
| 1.020 | -0.007 | -0.652 | 15.856 | -0.145 | -0.107 |
| -0.832 | -0.195 | 0.625 | -0.145 | 16.127 | 0.969 |
| -50.119 | 14.850 | -14.479 | -3.021 | 15.986 | 39.476 |
| 66.362 | 18.869 | 44.432 | 4.304 | -11.087 | -30.482 |
| 53.520 | 44.628 | 41.176 | 2.689 | -2.686 | -16.738 |
| 39.159 | 40.256 | 67.151 | -0.509 | -1.102 | -9.351 |
| 1.020 | -0.007 | -0.652 | 15.856 | -0.145 | -0.107 |
| 9.582 | 0.539 | 7.052 | 0.425 | 8.852 | -10.996 |
| 0.237 | -0.574 | -0.159 | -0.170 | 2.124 | 16.698 |
| 64.231 | 34.379 | 34.280 | 0.981 | 0.106 | 1.162 |
| 34.358 | 64.638 | 34.001 | 1.083 | -0.257 | -0.567 |
| 34.312 | 34.103 | 54.699 | -0.186 | 1.492 | 1.892 |
| -2.834 | 3.074 | -0.464 | 3.676 | 1.032 | 1.572 |
| -0.832 | -0.195 | 0.625 | -0.145 | 16.127 | 0.969 |
| 0.237 | -0.573 | -0.159 | -0.170 | 2.124 | 16.698 |
| 64.231 | 34.379 | 34.279 | 0.981 | 0.106 | 1.162 |
| 36.958 | 50.739 | 34.384 | 1.437 | -0.001 | -2.608 |
| 34.010 | 33.938 | 66.842 | -0.652 | 0.625 | -0.180 |
| 2.497 | 0.145 | 0.690 | 9.158 | -0.418 | -2.253 |
| -0.518 | 0.604 | 1.018 | -0.253 | 13.010 | 0.139 |
| 1.329 | -1.578 | 1.155 | -0.645 | 1.889 | 7.364 |
| -2105.014 | -2387.232 | -888.793 | -1031.910 | 330.125 | 593.275 |
| -103.875 | -396.360 | 46.371 | -324.067 | 40.015 | 127.261 |
| 33.964 | 35.090 | 65.892 | -2.109 | -0.463 | -0.304 |
| 9.926 | -271.897 | 5.539 | -268.450 | -16.867 | 23.863 |
| 0.190 | 0.420 | -0.463 | -0.376 | 15.764 | 0.411 |
| 10.505 | -269.595 | 7.415 | -282.330 | -15.748 | 37.517 |
| 50.299 | 305.770 | 27.134 | 282.656 | 17.318 | -21.447 |
| 26.786 | 331.466 | 27.805 | 280.115 | 16.605 | -24.095 |
| 25.179 | 304.926 | 53.307 | 280.307 | 15.755 | -24.026 |
| -9.515 | 266.485 | -10.099 | 296.720 | 15.987 | -23.492 |
| 0.190 | 0.420 | -0.463 | -0.376 | 15.764 | 0.411 |
| 0.859 | -0.217 | -0.351 | 0.272 | 0.967 | 17.012 |
| 60.987 | 9.660 | 35.736 | -28.190 | -0.752 | 4.599 |
| -176.524 | -204.918 | -56.596 | -107.965 | 32.181 | 59.257 |
| 33.964 | 35.090 | 65.892 | -2.109 | -0.463 | -0.304 |
| 0.220 | -2.514 | -2.109 | 16.753 | -0.376 | 0.218 |
| -212.595 | -242.597 | -98.337 | -103.752 | 33.414 | 59.793 |
| -431.273 | -392.350 | -134.032 | -161.987 | 93.860 | 185.395 |
| 64.630 | 35.245 | 34.029 | 0.116 | 0.936 | 2.629 |
| 204.165 | 235.833 | 91.036 | 67.327 | -33.011 | -54.830 |
| 33.964 | 35.090 | 65.892 | -2.109 | -0.463 | -0.304 |
| 0.220 | -2.514 | -2.109 | 16.753 | -0.376 | 0.218 |
| -0.700 | 27.058 | -1.454 | 27.830 | 14.411 | -2.142 |
| -0.187 | 26.516 | -1.185 | 28.515 | 2.322 | 11.469 |
| 9.728 | 6.303 | 24.795 | -14.462 | 3.147 | 11.134 |
| 29.252 | 38.791 | 36.770 | -7.481 | 0.592 | 3.656 |
| 27.962 | 27.364 | 44.162 | -6.805 | -1.744 | 4.320 |
| 0.220 | -2.514 | -2.109 | 16.753 | -0.376 | 0.218 |
| 0.370 | -2.562 | -0.594 | -3.264 | 12.608 | 0.464 |
| 0.859 | -0.217 | -0.351 | 0.272 | 0.967 | 17.012 |
| 86.654 | 68.824 | 48.591 | 12.826 | -10.151 | -11.066 |
| 45.740 | 58.305 | 36.686 | 0.237 | -1.916 | -4.188 |
| 52.119 | 54.786 | 38.987 | 3.776 | -7.911 | -5.061 |
| 1.794 | 1.565 | -2.743 | 15.311 | -0.907 | -1.123 |
| 0.176 | 2.836 | -0.752 | 2.405 | 12.913 | -0.013 |
| 0.686 | 2.296 | -0.482 | 3.092 | 0.841 | 13.570 |
| 150.209 | 56.826 | 74.349 | -0.158 | 2.694 | -13.547 |
| 33.561 | 66.561 | 33.555 | 0.987 | 1.056 | -0.251 |
| 33.037 | 33.435 | 63.881 | 2.109 | 0.088 | 1.314 |
| 0.174 | 0.076 | 2.109 | 16.009 | 1.341 | 0.836 |
| 404.587 | -37.242 | 25.116 | -113.963 | 15.573 | -64.981 |
| 582.384 | -228.115 | -32.471 | -403.381 | 44.374 | -104.915 |
| 63.846 | 33.561 | 33.214 | 0.352 | 0.530 | 1.234 |
| 33.561 | 66.561 | 33.555 | 0.987 | 1.056 | -0.251 |
| -368.754 | 73.754 | 27.109 | 117.838 | -6.961 | 67.287 |
| 0.174 | 0.076 | 2.109 | 16.009 | 1.341 | 0.836 |
| -796.459 | 410.173 | -86.009 | 557.831 | -45.001 | 127.443 |
| -0.921 | -0.242 | 1.319 | 0.912 | 1.119 | 16.108 |
| 63.846 | 33.561 | 33.214 | 0.352 | 0.530 | 1.234 |
| 75.048 | 50.721 | 38.219 | -11.351 | 1.722 | -6.469 |
| 33.037 | 33.435 | 63.881 | 2.109 | 0.088 | 1.314 |
| 0.174 | 0.076 | 2.109 | 16.009 | 1.341 | 0.836 |
| 40.099 | -2.910 | 2.828 | -10.071 | 8.993 | -5.485 |
| 39.530 | -3.621 | 3.669 | -11.069 | 2.325 | 3.413 |
| 50.243 | 32.058 | 30.664 | 0.104 | 0.114 | 2.483 |
| 25.221 | 59.462 | 30.640 | 0.798 | 0.720 | 1.298 |
| -4.260 | 39.423 | 49.393 | 13.943 | -0.377 | 7.790 |
| 0.174 | 0.076 | 2.109 | 16.009 | 1.341 | 0.836 |
| -0.291 | 0.980 | 0.088 | 1.341 | 15.736 | 0.223 |
| -9.877 | -2.397 | -2.866 | 0.778 | 1.056 | 14.864 |
| 60.392 | 34.514 | 34.992 | 0.087 | 0.378 | 0.885 |
| 33.561 | 66.562 | 33.555 | 0.987 | 1.056 | -0.251 |
| 35.455 | 34.727 | 59.220 | 2.423 | 0.355 | 0.930 |
| 0.174 | 0.076 | 2.109 | 16.009 | 1.341 | 0.836 |
| -0.291 | 0.980 | 0.088 | 1.341 | 15.736 | 0.223 |
| 3.021 | -0.189 | 1.439 | -0.655 | 1.657 | 9.477 |
| 63.846 | 33.561 | 33.214 | 0.352 | 0.530 | 1.234 |
| 28.609 | 54.529 | 35.666 | 3.751 | 0.729 | 1.247 |
| 33.580 | 34.248 | 58.518 | 2.421 | 0.303 | 1.230 |
| 0.174 | 0.076 | 2.109 | 16.009 | 1.341 | 0.836 |
| -7.970 | 5.255 | -0.100 | 6.948 | 6.039 | 2.372 |
| -0.921 | -0.242 | 1.319 | 0.912 | 1.119 | 16.108 |
| -97.207 | -162.986 | 2.579 | 190.144 | -294.442 | 203.293 |
| 34.682 | 64.412 | 35.236 | 0.911 | -0.492 | -1.582 |
| 36.283 | 34.808 | 71.279 | 0.933 | 3.178 | 0.529 |
| 1.657 | -10.460 | -11.604 | 26.618 | -64.158 | 29.609 |
| -148.071 | -208.264 | -33.397 | 188.895 | -335.430 | 246.424 |
| 0.526 | -10.088 | -12.658 | 17.428 | -64.827 | 36.982 |
| 71.212 | 34.293 | 36.629 | 0.418 | 1.256 | 1.663 |
| 21.286 | 50.443 | 42.915 | 0.223 | 25.426 | -8.292 |
| 197.236 | 247.085 | 79.513 | -188.468 | 337.206 | -249.055 |
| 0.350 | 0.534 | 0.933 | 16.933 | 0.553 | -0.618 |
| -0.440 | -6.238 | 13.556 | -2.841 | 61.641 | -16.193 |
| 0.465 | -1.582 | 0.591 | -0.490 | 0.572 | 17.122 |
| 68.592 | 36.409 | 36.984 | 0.874 | -1.625 | 2.371 |
| 38.334 | 39.542 | 39.099 | 1.999 | -8.712 | 4.958 |
| 39.288 | 37.546 | 56.076 | 2.497 | -3.915 | 2.605 |
| 2.196 | 1.371 | 0.131 | 14.684 | -1.922 | 0.069 |
| 0.229 | -0.620 | 3.178 | 0.553 | 17.937 | 0.344 |
| 0.465 | -1.582 | 0.591 | -0.490 | 0.572 | 17.122 |
| 52.366 | 40.651 | 41.824 | -0.220 | 9.983 | -3.021 |
| 34.682 | 64.412 | 35.236 | 0.911 | -0.492 | -1.582 |
| 36.283 | 34.808 | 71.279 | 0.933 | 3.178 | 0.529 |
| 0.350 | 0.534 | 0.933 | 16.933 | 0.553 | -0.618 |
| 8.179 | 3.105 | 4.052 | -5.748 | 12.535 | -11.439 |
| 0.782 | -1.812 | 1.197 | -0.754 | 5.227 | 9.340 |
| 65.879 | 35.927 | 37.949 | 0.971 | 0.559 | 1.790 |
| 36.310 | 43.087 | 38.476 | -1.279 | -3.012 | 3.699 |
| 36.283 | 34.808 | 71.279 | 0.933 | 3.178 | 0.529 |
| 1.010 | 0.464 | 0.701 | 14.115 | 0.378 | -0.518 |
| 0.239 | -0.475 | 2.606 | 0.736 | 12.482 | 0.339 |
| 1.019 | 0.279 | -0.724 | -0.086 | -0.767 | 7.233 |
| 66.082 | 35.511 | 37.978 | 0.901 | 1.268 | 1.611 |
| 36.967 | 54.270 | 37.830 | 0.190 | 0.128 | -1.358 |
| 39.789 | 38.341 | 57.729 | 0.547 | 3.339 | -1.106 |
| 1.417 | -0.091 | 0.837 | 11.561 | 1.196 | -0.688 |
| 0.292 | -0.557 | 2.805 | 0.663 | 13.267 | -0.016 |
| 0.857 | -0.116 | -0.202 | -0.431 | 0.498 | 7.590 |
| 35.068 | 84.010 | 7.122 | 0.000 | 0.000 | 0.000 |
| 34.671 | 73.294 | 29.608 | 0.000 | 0.000 | 0.000 |
| 15.760 | 88.801 | 7.587 | -0.000 | -0.000 | -0.000 |
| -17.274 | 46.774 | -21.895 | 11.824 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 25.250 | 0.000 |
| -16.700 | 45.220 | -21.168 | -0.000 | -0.000 | 5.910 |
| 80.254 | -15.183 | 54.268 | -0.000 | 0.000 | -0.000 |
| 21.589 | 63.420 | 32.836 | -0.000 | 0.000 | -0.000 |
| 35.520 | 7.586 | 44.751 | 0.000 | -0.000 | 0.000 |
| 17.274 | -46.774 | 21.895 | 11.824 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 25.250 | -0.000 |
| 16.700 | -45.220 | 21.168 | -0.000 | -0.000 | 5.910 |
| 71.828 | 21.049 | 37.720 | 0.000 | 0.000 | 0.000 |
| 33.142 | 41.274 | 43.484 | -0.000 | 0.000 | -0.000 |
| 25.079 | 52.012 | 24.452 | 0.000 | 0.000 | -0.000 |
| -3.339 | 9.040 | -4.232 | 3.573 | 0.000 | 0.000 |
| -1.413 | 4.252 | -1.977 | 0.000 | 18.012 | 0.000 |
| -0.968 | 4.049 | -1.852 | -0.000 | 0.000 | -0.191 |
| 50.204 | 44.155 | 25.813 | 0.000 | 0.000 | 0.000 |
| 21.589 | 63.420 | 32.836 | 0.000 | 0.000 | -0.000 |
| 32.453 | 42.250 | 29.336 | 0.000 | -0.000 | 0.000 |
| 0.938 | -3.458 | 1.590 | 0.183 | 0.000 | 0.000 |
| 1.413 | -4.252 | 1.977 | -0.000 | 18.012 | 0.000 |
| 0.968 | -4.049 | 1.852 | -0.000 | -0.000 | -0.191 |
| 55.859 | 43.316 | 26.651 | -0.000 | 0.000 | -0.000 |
| 44.069 | 33.311 | 47.877 | 0.000 | 0.000 | -0.000 |
| 31.470 | 45.907 | 27.655 | 0.000 | 0.000 | -0.000 |
| -0.375 | 1.015 | -0.475 | 1.465 | 0.000 | 0.000 |
| -0.201 | 0.545 | -0.255 | 0.000 | 18.035 | 0.000 |
| -0.097 | 0.405 | -0.185 | -0.000 | -0.000 | -0.191 |
| 71.828 | 21.049 | 37.720 | 0.000 | 0.000 | -0.000 |
| 34.573 | 37.035 | 45.457 | -0.000 | -0.000 | 0.000 |
| 31.718 | 45.154 | 28.005 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.120 | 0.052 | 0.236 | -0.000 | -0.000 |
| 0.201 | -0.545 | 0.255 | -0.000 | 18.035 | -0.000 |
| 0.097 | -0.405 | 0.185 | -0.000 | -0.000 | -0.191 |
| 66.276 | 28.909 | 28.909 | -0.000 | -0.000 | -0.000 |
| 28.909 | 66.276 | 28.909 | 0.000 | -0.000 | 0.000 |
| 28.909 | 28.909 | 66.276 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 8.684 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 8.684 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 8.684 |
| -717.006 | -742.884 | -742.884 | 0.000 | 0.000 | 0.000 |
| -742.884 | -717.006 | -742.884 | 0.000 | 0.000 | 0.000 |
| -815.839 | -815.839 | -783.013 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 8.684 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 8.684 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 8.684 |
| 66.276 | 28.909 | 28.909 | -0.000 | -0.000 | -0.000 |
| 28.909 | 66.276 | 28.909 | -0.000 | -0.000 | -0.000 |
| 28.909 | 28.909 | 66.276 | -0.000 | -0.000 | -0.000 |
| 58.615 | 58.615 | 58.615 | -9.051 | -0.000 | -0.000 |
| 58.615 | 58.615 | 58.615 | -0.000 | -9.051 | -0.000 |
| 58.615 | 58.615 | 58.615 | -0.000 | -0.000 | -9.051 |
| 66.276 | 28.909 | 28.909 | 0.000 | 0.000 | 0.000 |
| 28.909 | 66.276 | 28.909 | 0.000 | 0.000 | -0.000 |
| 28.909 | 28.909 | 66.276 | 0.000 | 0.000 | 0.000 |
| -58.615 | -58.615 | -58.615 | -9.051 | 0.000 | 0.000 |
| -58.615 | -58.615 | -58.615 | 0.000 | -9.051 | 0.000 |
| -58.615 | -58.615 | -58.615 | 0.000 | 0.000 | -9.051 |
| 66.277 | 28.909 | 28.909 | 0.000 | -0.000 | -0.000 |
| 28.909 | 66.277 | 28.909 | -0.000 | -0.000 | 0.000 |
| 28.909 | 28.909 | 66.277 | 0.000 | -0.000 | -0.000 |
| 2.868 | 2.869 | 2.869 | 0.005 | -0.000 | -0.000 |
| 2.869 | 2.868 | 2.869 | -0.000 | 0.005 | -0.000 |
| 2.869 | 2.869 | 2.868 | -0.000 | -0.000 | 0.005 |
| 49.774 | 24.611 | 24.611 | 0.000 | 0.000 | 0.000 |
| 24.611 | 49.774 | 24.611 | 0.000 | 0.000 | 0.000 |
| 24.611 | 24.611 | 49.774 | 0.000 | 0.000 | 0.000 |
| -2.868 | -2.869 | -2.869 | 0.005 | 0.000 | 0.000 |
| -2.869 | -2.868 | -2.869 | 0.000 | 0.005 | 0.000 |
| -2.869 | -2.869 | -2.868 | 0.000 | 0.000 | 0.005 |
| 91.846 | 82.130 | 82.130 | 0.000 | 0.000 | 0.000 |
| 82.130 | 91.846 | 82.130 | 0.000 | 0.000 | 0.000 |
| 82.130 | 82.130 | 91.846 | 0.000 | 0.000 | 0.000 |
| 42.567 | 42.567 | 42.567 | 5.285 | 0.000 | 0.000 |
| 42.567 | 42.567 | 42.567 | 0.000 | 5.285 | 0.000 |
| 42.567 | 42.567 | 42.567 | 0.000 | 0.000 | 5.285 |
| 55.622 | 36.581 | 36.581 | 0.000 | 0.000 | -0.000 |
| 36.581 | 55.622 | 36.581 | -0.000 | 0.000 | 0.000 |
| 36.581 | 36.581 | 55.622 | 0.000 | 0.000 | 0.000 |
| -42.567 | -42.567 | -42.567 | 5.285 | -0.000 | -0.000 |
| -42.567 | -42.567 | -42.567 | -0.000 | 5.285 | -0.000 |
| -42.567 | -42.567 | -42.567 | -0.000 | -0.000 | 5.285 |
| 53.514 | 43.477 | 43.477 | 0.000 | 0.000 | 0.000 |
| 43.477 | 53.514 | 43.477 | 0.000 | 0.000 | 0.000 |
| 43.477 | 43.477 | 53.514 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 14.920 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 14.920 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 14.920 |
| 41.000 | 34.013 | 34.013 | 0.000 | 0.000 | 0.000 |
| 34.013 | 41.000 | 34.013 | 0.000 | 0.000 | 0.000 |
| 34.013 | 34.013 | 41.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 14.920 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 14.920 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 14.920 |
| 55.621 | 36.581 | 36.581 | 0.000 | -0.000 | -0.000 |
| 36.581 | 55.621 | 36.581 | -0.000 | 0.000 | -0.000 |
| 36.581 | 36.581 | 55.621 | -0.000 | -0.000 | 0.000 |
| 0.395 | 0.395 | 0.395 | 7.817 | -0.000 | -0.000 |
| 0.395 | 0.395 | 0.395 | -0.000 | 7.817 | -0.000 |
| 0.395 | 0.395 | 0.395 | -0.000 | -0.000 | 7.817 |
| 55.622 | 36.581 | 36.581 | -0.000 | -0.000 | 0.000 |
| 36.581 | 55.622 | 36.581 | 0.000 | -0.000 | -0.000 |
| 36.581 | 36.581 | 55.622 | -0.000 | 0.000 | -0.000 |
| -0.395 | -0.395 | -0.395 | 7.817 | 0.000 | 0.000 |
| -0.395 | -0.395 | -0.395 | 0.000 | 7.817 | 0.000 |
| -0.395 | -0.395 | -0.395 | 0.000 | 0.000 | 7.817 |
| 56.554 | 37.823 | 37.823 | -0.000 | -0.000 | 0.000 |
| 37.823 | 56.554 | 37.823 | 0.000 | 0.000 | 0.000 |
| 37.823 | 37.823 | 56.554 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 14.696 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 14.696 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 14.696 |
| 57.644 | 45.265 | 45.265 | 0.003 | 0.003 | 0.003 |
| 45.265 | 57.644 | 45.265 | 0.003 | 0.003 | 0.003 |
| 45.265 | 45.265 | 57.644 | 0.003 | 0.003 | 0.003 |
| -0.000 | 0.000 | -0.000 | 14.696 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 14.696 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 14.696 |
| 49.378 | 40.340 | 40.340 | -0.000 | -0.000 | -0.000 |
| 40.340 | 49.378 | 40.340 | -0.000 | -0.000 | -0.000 |
| 40.340 | 40.340 | 49.378 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 14.696 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 14.696 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 14.696 |
| 56.554 | 37.823 | 37.823 | -0.000 | 0.000 | 0.000 |
| 37.823 | 56.554 | 37.823 | -0.000 | -0.000 | -0.000 |
| 37.823 | 37.823 | 56.554 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 14.696 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 14.696 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 14.696 |
| 56.555 | 37.823 | 37.823 | 0.000 | -0.000 | 0.000 |
| 37.823 | 56.555 | 37.823 | -0.000 | -0.000 | 0.000 |
| 37.823 | 37.823 | 56.555 | 0.000 | 0.000 | 0.000 |
| -0.053 | -0.053 | -0.053 | 8.385 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 14.696 | 0.000 |
| -0.053 | -0.053 | -0.053 | -0.000 | -0.000 | 8.385 |
| 56.553 | 37.822 | 37.822 | -0.000 | 0.000 | -0.000 |
| 37.822 | 56.553 | 37.822 | -0.000 | 0.000 | -0.000 |
| 37.822 | 37.822 | 56.553 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 14.696 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 14.696 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 14.696 |
| 62.901 | 37.350 | 27.425 | -0.000 | 0.000 | 0.000 |
| 37.286 | 72.070 | 13.958 | -0.000 | 0.000 | 0.000 |
| 28.414 | 12.741 | 86.280 | -0.000 | 0.000 | 0.000 |
| -0.008 | 0.008 | 0.035 | -3.641 | -0.000 | 0.000 |
| -0.008 | 0.008 | 0.035 | -0.000 | 12.269 | 0.000 |
| -0.001 | 0.001 | 0.006 | 0.000 | -0.000 | 20.032 |
| 62.901 | 37.350 | 27.425 | -0.000 | -0.000 | -0.000 |
| 37.558 | 71.799 | 12.817 | 0.000 | -0.000 | -0.000 |
| 28.417 | 12.738 | 86.268 | 0.000 | -0.000 | -0.000 |
| 0.008 | -0.008 | -0.035 | -3.641 | -0.000 | -0.000 |
| 0.008 | -0.008 | -0.035 | 0.000 | 12.269 | -0.000 |
| 0.001 | -0.001 | -0.006 | 0.000 | 0.000 | 20.032 |
| 62.901 | 37.350 | 27.425 | -0.000 | 0.000 | 0.000 |
| 37.286 | 72.070 | 13.958 | -0.000 | 0.000 | -0.000 |
| 27.279 | 13.875 | 91.052 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.064 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 12.611 | 0.000 |
| -0.000 | 0.000 | 0.001 | -0.000 | 0.000 | 20.032 |
| 62.630 | 37.620 | 28.562 | 0.000 | -0.000 | 0.000 |
| 37.286 | 72.070 | 13.958 | 0.000 | -0.000 | -0.000 |
| 27.279 | 13.875 | 91.052 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.064 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 12.611 | -0.000 |
| 0.000 | -0.000 | -0.001 | 0.000 | 0.000 | 20.032 |
| 62.631 | 37.620 | 28.562 | -0.000 | -0.000 | 0.000 |
| 37.556 | 71.801 | 12.822 | -0.000 | 0.000 | 0.000 |
| 28.415 | 12.739 | 86.275 | -0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.002 | -7.376 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 12.611 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 20.032 |
| 62.630 | 37.620 | 28.562 | 0.000 | 0.000 | -0.000 |
| 37.287 | 72.070 | 13.958 | 0.000 | -0.000 | -0.000 |
| 28.416 | 12.739 | 86.274 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.002 | -7.376 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 12.611 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 20.032 |
| 55.176 | 25.882 | 8.017 | -0.000 | 0.000 | 0.000 |
| 25.882 | 55.176 | 8.017 | -0.000 | 0.000 | -0.000 |
| 8.017 | 8.017 | 40.660 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.034 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.034 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -7.309 |
| 55.176 | 25.882 | 8.017 | 0.000 | -0.000 | 0.000 |
| 25.882 | 55.176 | 8.017 | 0.000 | -0.000 | -0.000 |
| 8.017 | 8.017 | 40.660 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.034 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.034 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | -7.309 |
| 33.172 | 47.886 | 8.017 | 0.000 | 0.000 | 0.000 |
| 47.886 | 33.173 | 8.017 | -0.000 | -0.000 | 0.000 |
| 8.017 | 8.017 | 40.660 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.034 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.034 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 14.647 |
| 33.221 | 47.838 | 8.017 | -0.000 | -0.000 | 0.000 |
| 25.882 | 55.176 | 8.017 | 0.000 | -0.000 | -0.000 |
| 8.017 | 8.017 | 40.660 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.034 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.034 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 14.647 |
| 32.882 | 48.176 | 8.017 | 0.000 | 0.000 | -0.000 |
| 47.887 | 33.172 | 8.017 | 0.000 | 0.000 | 0.000 |
| 8.017 | 8.017 | 40.661 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.034 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.034 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -7.309 |
| 33.220 | 47.838 | 8.017 | -0.000 | -0.000 | 0.000 |
| 47.837 | 33.221 | 8.017 | -0.000 | -0.000 | 0.000 |
| 8.017 | 8.017 | 40.660 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.034 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.034 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -7.309 |
| 51.023 | 30.766 | 43.624 | 0.141 | 0.000 | -0.000 |
| 30.723 | 50.853 | 43.091 | -0.231 | -0.000 | -0.000 |
| 43.624 | 43.624 | 84.621 | -0.000 | -0.000 | -0.000 |
| 0.398 | -0.394 | 0.009 | 22.318 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 22.683 | 0.141 |
| 0.002 | 0.002 | 0.009 | -0.000 | 0.396 | 9.949 |
| 51.023 | 30.766 | 43.624 | 0.141 | -0.000 | -0.000 |
| 30.766 | 51.023 | 43.624 | -0.141 | -0.000 | -0.000 |
| 43.237 | 43.237 | 82.679 | 0.000 | -0.000 | 0.000 |
| 0.141 | -0.141 | -0.000 | 22.683 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 22.683 | 0.141 |
| -0.002 | -0.002 | -0.009 | 0.000 | 0.396 | 9.949 |
| 50.766 | 30.868 | 43.237 | 0.396 | 0.000 | 0.000 |
| 30.722 | 50.853 | 43.089 | -0.231 | -0.000 | -0.000 |
| 43.624 | 43.624 | 84.621 | -0.000 | 0.000 | 0.000 |
| 0.396 | -0.396 | 0.001 | 22.318 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 22.683 | 0.141 |
| 0.000 | 0.000 | 0.001 | -0.000 | 0.396 | 9.949 |
| 51.023 | 30.766 | 43.624 | 0.141 | -0.000 | 0.000 |
| 30.722 | 50.852 | 43.089 | -0.231 | -0.000 | 0.000 |
| 43.237 | 43.237 | 82.681 | -0.000 | -0.000 | 0.000 |
| 0.396 | -0.396 | -0.001 | 22.318 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 22.683 | 0.141 |
| -0.000 | -0.000 | -0.001 | 0.000 | 0.396 | 9.949 |
| 50.853 | 30.723 | 43.089 | 0.231 | 0.000 | -0.000 |
| 30.766 | 51.023 | 43.625 | -0.141 | 0.000 | 0.000 |
| 43.238 | 43.238 | 82.682 | -0.000 | 0.000 | -0.000 |
| 0.396 | -0.396 | 0.000 | 22.318 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 22.318 | 0.396 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.141 | 10.128 |
| 51.022 | 30.766 | 43.624 | 0.141 | -0.000 | 0.000 |
| 30.766 | 51.022 | 43.624 | -0.141 | -0.000 | 0.000 |
| 43.624 | 43.624 | 84.620 | -0.000 | -0.000 | 0.000 |
| 0.396 | -0.396 | -0.000 | 22.318 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 22.318 | 0.396 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.141 | 10.128 |
| 51.057 | 30.822 | 44.008 | 0.187 | 0.000 | -0.000 |
| 32.862 | 52.996 | 53.939 | -0.255 | -0.000 | -0.000 |
| 45.636 | 45.636 | 93.827 | 0.000 | 0.000 | -0.000 |
| 0.187 | -0.187 | 0.000 | 22.899 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 22.899 | 0.187 |
| 2.069 | 2.069 | 10.330 | 0.000 | 0.255 | 10.067 |
| 51.057 | 30.822 | 44.008 | 0.187 | -0.000 | -0.000 |
| 30.822 | 51.057 | 44.008 | -0.187 | -0.000 | 0.000 |
| 43.967 | 43.967 | 85.497 | 0.000 | -0.000 | 0.000 |
| -1.816 | -2.325 | -10.336 | 22.807 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 22.899 | 0.187 |
| -2.069 | -2.069 | -10.330 | -0.000 | 0.255 | 10.067 |
| 50.989 | 31.144 | 44.641 | 0.451 | 0.000 | -0.000 |
| 31.144 | 50.989 | 44.641 | -0.451 | -0.000 | -0.000 |
| 43.775 | 43.775 | 84.537 | 0.000 | 0.000 | 0.000 |
| 0.658 | -0.244 | 1.032 | 22.541 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 22.899 | 0.187 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.187 | 10.117 |
| 50.575 | 30.730 | 42.576 | 0.451 | -0.000 | 0.000 |
| 30.577 | 50.708 | 42.525 | -0.257 | -0.000 | 0.000 |
| 43.967 | 43.967 | 85.497 | 0.000 | -0.000 | 0.000 |
| 0.031 | -0.495 | -1.158 | 22.796 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 22.899 | 0.187 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.187 | 10.117 |
| 50.877 | 30.807 | 43.519 | 0.299 | -0.000 | -0.000 |
| 30.823 | 51.057 | 44.008 | -0.187 | 0.000 | -0.000 |
| 43.968 | 43.968 | 85.498 | 0.000 | 0.000 | 0.000 |
| 0.484 | -0.345 | 0.348 | 22.590 | 0.000 | -0.000 |
| 0.070 | 0.070 | 0.348 | 0.000 | 22.590 | 0.415 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.187 | 10.117 |
| 51.056 | 30.822 | 44.008 | 0.187 | -0.000 | -0.000 |
| 30.758 | 50.866 | 43.371 | -0.273 | -0.000 | 0.000 |
| 43.547 | 43.547 | 83.400 | 0.000 | -0.000 | -0.000 |
| 0.345 | -0.484 | -0.348 | 22.590 | -0.000 | -0.000 |
| -0.070 | -0.070 | -0.348 | -0.000 | 22.590 | 0.415 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.187 | 10.117 |
| 390.101 | 182.905 | 153.489 | -0.000 | -0.000 | -0.000 |
| 182.905 | 390.101 | 153.489 | 0.000 | 0.000 | -0.000 |
| 3031.106 | 3031.106 | 7954.932 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 102.072 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 102.072 | 0.000 |
| 2804.734 | 2804.734 | 7652.194 | -0.000 | -0.000 | 83.157 |
| -929.313 | -1117.637 | -7839.011 | 0.000 | 0.000 | -0.000 |
| -2620.035 | -2453.720 | -7498.563 | 0.000 | 0.000 | 0.000 |
| -1148.267 | -1148.267 | -7710.601 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 102.072 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 102.072 | 0.000 |
| -2804.734 | -2804.734 | -7652.194 | -0.000 | 0.000 | 83.157 |
| 500.576 | 312.152 | 948.175 | 0.000 | -0.000 | -0.000 |
| 312.233 | 500.645 | 948.674 | 0.000 | -0.000 | -0.000 |
| 281.586 | 281.586 | 1076.868 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 102.072 | -0.000 | -0.000 |
| 280.468 | 280.468 | 765.204 | 0.000 | 91.012 | -0.000 |
| 130.006 | 130.006 | 798.989 | -0.000 | -0.000 | 94.165 |
| 240.434 | 52.191 | -649.492 | 0.000 | 0.000 | 0.000 |
| 52.191 | 240.434 | -649.492 | -0.000 | 0.000 | -0.000 |
| -137.517 | -137.517 | -491.428 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 102.072 | 0.000 | 0.000 |
| -280.468 | -280.468 | -765.204 | 0.000 | 91.012 | 0.000 |
| -130.006 | -130.006 | -798.989 | -0.000 | 0.000 | 94.165 |
| 390.098 | 182.901 | 153.487 | 0.000 | -0.000 | -0.000 |
| 182.901 | 390.097 | 153.487 | -0.000 | -0.000 | -0.000 |
| 151.336 | 151.336 | 297.747 | 0.000 | -0.000 | -0.000 |
| 13.077 | 13.077 | 80.369 | 95.782 | -0.000 | -0.000 |
| 13.077 | 13.077 | 80.369 | -0.000 | 95.782 | -0.000 |
| 13.209 | 13.209 | 81.180 | -0.000 | -0.000 | 94.014 |
| 355.903 | 168.886 | 67.696 | -0.000 | 0.000 | 0.000 |
| 156.404 | 322.434 | 76.448 | -0.000 | 0.000 | 0.000 |
| 151.337 | 151.337 | 297.745 | 0.000 | 0.000 | 0.000 |
| -13.077 | -13.077 | -80.369 | 95.782 | 0.000 | 0.000 |
| -13.077 | -13.077 | -80.369 | 0.000 | 95.782 | 0.000 |
| -13.209 | -13.209 | -81.180 | 0.000 | 0.000 | 94.014 |
| 394.464 | 181.956 | 149.974 | 0.000 | 0.000 | 0.000 |
| 4104.114 | 4293.990 | 6989.217 | -0.000 | 0.000 | -0.000 |
| 6489.809 | 6489.809 | 5083.001 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 102.621 | 0.000 | -0.000 |
| 6355.034 | 6355.034 | 4746.145 | 0.000 | 91.666 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 106.254 |
| -3555.122 | -3744.959 | -6695.029 | -0.000 | -0.000 | 0.000 |
| -6150.996 | -5979.261 | -4678.471 | -0.000 | 0.000 | 0.000 |
| 149.974 | 149.974 | 275.949 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 102.621 | 0.000 | 0.000 |
| -6355.034 | -6355.034 | -4746.145 | -0.000 | 91.666 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 106.254 |
| 394.464 | 181.956 | 149.974 | -0.000 | 0.000 | -0.000 |
| 820.448 | 996.188 | 626.871 | 0.000 | 0.000 | -0.000 |
| 550.891 | 550.891 | 939.797 | 0.000 | 0.000 | -0.000 |
| 635.504 | 635.504 | 474.603 | 91.667 | 0.000 | -0.000 |
| 635.504 | 635.504 | 474.603 | 0.000 | 91.667 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 106.254 |
| -23.245 | -212.908 | -537.165 | -0.000 | -0.000 | -0.000 |
| -446.852 | -275.209 | -331.344 | -0.000 | -0.000 | -0.000 |
| 149.974 | 149.974 | 275.949 | -0.000 | -0.000 | 0.000 |
| -635.504 | -635.504 | -474.603 | 91.667 | -0.000 | 0.000 |
| -635.504 | -635.504 | -474.603 | -0.000 | 91.666 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 106.254 |
| 410.767 | 219.342 | 216.331 | -0.000 | 0.000 | -0.000 |
| 181.956 | 394.465 | 149.974 | 0.000 | 0.000 | -0.000 |
| 199.794 | 199.794 | 321.844 | 0.000 | 0.000 | -0.000 |
| 63.561 | 63.561 | 47.347 | 91.669 | 0.000 | -0.000 |
| 63.561 | 63.561 | 47.347 | 0.000 | 91.669 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 106.254 |
| 394.463 | 181.956 | 149.974 | -0.000 | -0.000 | -0.000 |
| 181.956 | 394.463 | 149.974 | -0.000 | 0.000 | 0.000 |
| 149.974 | 149.974 | 275.948 | 0.000 | -0.000 | 0.000 |
| -63.561 | -63.561 | -47.347 | 91.669 | -0.000 | 0.000 |
| -63.561 | -63.561 | -47.347 | 0.000 | 91.669 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 106.254 |
| 389.349 | 182.094 | 153.077 | 0.000 | -0.000 | 0.000 |
| 571.041 | 740.240 | -200.079 | 0.000 | 0.000 | -0.000 |
| 150.946 | 150.946 | 297.399 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 102.091 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 102.091 | -0.000 |
| 387.681 | 387.681 | -351.728 | 0.000 | 0.000 | 84.666 |
| 389.349 | 182.094 | 153.077 | 0.000 | 0.000 | 0.000 |
| 182.094 | 389.349 | 153.077 | 0.000 | -0.000 | 0.000 |
| 150.947 | 150.947 | 297.399 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 102.091 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 102.091 | -0.000 |
| -387.681 | -387.681 | 351.728 | 0.000 | -0.000 | 84.666 |
| 389.349 | 182.093 | 153.077 | 0.000 | -0.000 | -0.000 |
| 222.181 | 390.121 | 116.418 | 0.000 | 0.000 | -0.000 |
| 185.916 | 185.916 | 251.608 | -0.000 | 0.000 | -0.000 |
| 34.283 | 34.283 | -22.882 | 91.727 | -0.000 | -0.000 |
| 38.460 | 38.460 | -34.893 | -0.000 | 89.625 | 0.000 |
| 38.865 | 38.865 | -35.261 | -0.000 | 0.000 | 84.619 |
| 389.349 | 182.094 | 153.078 | -0.000 | 0.000 | -0.000 |
| 144.384 | 314.847 | 187.019 | 0.000 | -0.000 | 0.000 |
| 120.335 | 120.335 | 302.201 | -0.000 | -0.000 | 0.000 |
| -34.283 | -34.283 | 22.882 | 91.727 | -0.000 | 0.000 |
| -38.460 | -38.460 | 34.893 | 0.000 | 89.625 | 0.000 |
| -38.865 | -38.865 | 35.261 | -0.000 | -0.000 | 84.619 |
| 351.804 | 188.894 | 150.612 | 0.000 | 0.000 | -0.000 |
| 182.090 | 389.346 | 153.075 | -0.000 | -0.000 | -0.000 |
| 154.171 | 154.171 | 273.678 | -0.000 | 0.000 | -0.000 |
| 3.115 | 3.115 | -2.993 | 91.492 | 0.000 | 0.000 |
| 3.507 | 3.507 | -2.071 | -0.000 | 91.733 | -0.000 |
| 4.109 | 4.109 | -3.728 | 0.000 | 0.000 | 83.531 |
| 389.352 | 182.098 | 153.079 | -0.000 | 0.000 | -0.000 |
| 182.098 | 389.352 | 153.079 | 0.000 | -0.000 | 0.000 |
| 150.947 | 150.947 | 297.398 | -0.000 | -0.000 | 0.000 |
| -3.115 | -3.115 | 2.993 | 91.492 | 0.000 | -0.000 |
| -3.507 | -3.507 | 2.071 | 0.000 | 91.733 | 0.000 |
| -4.109 | -4.109 | 3.728 | -0.000 | -0.000 | 83.531 |
| 187.126 | 85.266 | 85.266 | -0.000 | 0.000 | 0.000 |
| 85.266 | 187.126 | 85.266 | -0.000 | -0.000 | -0.000 |
| 85.266 | 85.266 | 187.126 | -0.000 | 0.000 | -0.000 |
| -97.045 | -97.045 | -97.045 | 54.433 | -0.000 | -0.000 |
| -97.045 | -97.045 | -97.045 | -0.000 | 54.433 | 0.000 |
| -97.045 | -97.045 | -97.045 | -0.000 | 0.000 | 54.433 |
| 187.126 | 85.266 | 85.266 | 0.000 | 0.000 | 0.000 |
| 85.266 | 187.126 | 85.266 | 0.000 | 0.000 | -0.000 |
| 85.266 | 85.266 | 187.126 | 0.000 | -0.000 | -0.000 |
| 97.045 | 97.045 | 97.045 | 54.433 | 0.000 | -0.000 |
| 97.045 | 97.045 | 97.045 | 0.000 | 54.433 | 0.000 |
| 97.045 | 97.045 | 97.045 | 0.000 | -0.000 | 54.433 |
| 187.127 | 85.266 | 85.266 | -0.000 | 0.000 | -0.000 |
| 85.266 | 187.127 | 85.266 | -0.000 | -0.000 | 0.000 |
| 85.266 | 85.266 | 187.127 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 56.095 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 56.095 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 56.095 |
| 187.126 | 85.266 | 85.266 | 0.000 | -0.000 | -0.000 |
| 85.266 | 187.126 | 85.266 | 0.000 | 0.000 | -0.000 |
| 85.266 | 85.266 | 187.126 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 56.095 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 56.095 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 56.095 |
| 169.624 | 81.836 | 81.836 | -0.000 | 0.000 | 0.000 |
| 85.266 | 187.128 | 85.266 | -0.000 | -0.000 | -0.000 |
| 81.836 | 81.836 | 169.624 | -0.000 | 0.000 | 0.000 |
| -1.001 | -1.001 | -1.001 | 52.994 | -0.000 | 0.000 |
| -1.001 | -1.001 | -1.001 | -0.000 | 52.994 | -0.000 |
| -1.001 | -1.001 | -1.001 | -0.000 | -0.000 | 52.994 |
| 187.125 | 85.265 | 85.265 | 0.000 | -0.000 | -0.000 |
| 85.265 | 187.125 | 85.265 | 0.000 | -0.000 | 0.000 |
| 85.265 | 85.265 | 187.125 | 0.000 | -0.000 | -0.000 |
| 1.001 | 1.001 | 1.001 | 52.994 | -0.000 | 0.000 |
| 1.001 | 1.001 | 1.001 | 0.000 | 52.994 | -0.000 |
| 1.001 | 1.001 | 1.001 | 0.000 | -0.000 | 52.994 |
| 192.583 | 89.289 | 89.289 | 0.000 | 0.000 | -0.000 |
| 89.289 | 192.583 | 89.289 | 0.000 | 0.000 | -0.000 |
| 89.289 | 89.289 | 192.583 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 56.476 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 56.476 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 56.476 |
| 186.968 | 103.833 | 103.833 | 0.000 | 0.000 | -0.000 |
| 103.833 | 186.968 | 103.833 | -0.000 | -0.000 | 0.000 |
| 103.833 | 103.833 | 186.968 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 56.476 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 56.476 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 56.476 |
| 192.584 | 89.290 | 89.290 | 0.000 | 0.000 | 0.000 |
| 89.290 | 192.584 | 89.290 | 0.000 | 0.000 | -0.000 |
| 89.290 | 89.290 | 192.584 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 56.476 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 56.476 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 56.476 |
| 170.380 | 93.257 | 93.257 | -0.000 | 0.000 | 0.000 |
| 93.237 | 170.405 | 93.237 | -0.000 | 0.000 | 0.000 |
| 93.237 | 93.237 | 170.405 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 56.476 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 56.476 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 56.476 |
| 192.586 | 89.291 | 89.291 | -0.000 | -0.000 | -0.000 |
| 89.291 | 192.586 | 89.291 | 0.000 | -0.000 | -0.000 |
| 89.291 | 89.291 | 192.586 | 0.000 | 0.000 | 0.000 |
| -0.329 | -0.329 | -0.329 | 53.418 | -0.000 | -0.000 |
| -0.329 | -0.329 | -0.329 | -0.000 | 53.418 | 0.000 |
| -0.329 | -0.329 | -0.329 | 0.000 | 0.000 | 53.418 |
| 176.232 | 86.326 | 86.326 | -0.000 | -0.000 | 0.000 |
| 92.037 | 169.205 | 92.037 | -0.000 | 0.000 | -0.000 |
| 86.326 | 86.326 | 176.232 | -0.000 | 0.000 | 0.000 |
| 0.329 | 0.329 | 0.329 | 53.418 | -0.000 | 0.000 |
| 0.329 | 0.329 | 0.329 | 0.000 | 53.418 | 0.000 |
| 0.329 | 0.329 | 0.329 | 0.000 | 0.000 | 53.418 |
| 187.000 | 85.202 | 85.202 | 0.000 | -0.000 | 0.000 |
| 85.202 | 187.000 | 85.202 | -0.000 | -0.000 | 0.000 |
| 85.202 | 85.202 | 187.000 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 56.075 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 56.075 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 56.075 |
| 171.289 | 81.710 | 81.710 | 0.000 | 0.000 | -0.000 |
| 81.710 | 171.289 | 81.710 | 0.000 | 0.000 | -0.000 |
| 81.710 | 81.710 | 171.289 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 56.075 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 56.075 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 56.075 |
| 163.854 | 87.203 | 87.203 | 0.000 | -0.000 | -0.000 |
| 87.203 | 163.854 | 87.203 | -0.000 | 0.000 | -0.000 |
| 87.203 | 87.203 | 163.854 | -0.000 | -0.000 | -0.000 |
| 0.313 | 0.313 | 0.313 | 53.122 | -0.000 | -0.000 |
| 0.313 | 0.313 | 0.313 | -0.000 | 53.122 | 0.000 |
| 0.313 | 0.313 | 0.313 | -0.000 | -0.000 | 53.122 |
| 170.765 | 82.460 | 82.460 | 0.000 | 0.000 | 0.000 |
| 82.460 | 170.765 | 82.460 | 0.000 | 0.000 | -0.000 |
| 82.460 | 82.460 | 170.765 | 0.000 | 0.000 | 0.000 |
| -0.313 | -0.313 | -0.313 | 53.122 | -0.000 | -0.000 |
| -0.313 | -0.313 | -0.313 | -0.000 | 53.122 | -0.000 |
| -0.313 | -0.313 | -0.313 | 0.000 | 0.000 | 53.122 |
| 170.653 | 82.557 | 82.557 | -0.000 | -0.000 | 0.000 |
| 82.557 | 170.653 | 82.557 | -0.000 | 0.000 | -0.000 |
| 82.557 | 82.557 | 170.653 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 56.075 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 56.075 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 56.075 |
| 170.671 | 82.538 | 82.538 | 0.000 | 0.000 | 0.000 |
| 82.538 | 170.671 | 82.538 | 0.000 | 0.000 | -0.000 |
| 82.538 | 82.538 | 170.671 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 56.075 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 56.075 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 56.075 |
| -420.474 | -535.751 | -535.751 | 0.000 | -0.000 | -0.000 |
| -535.751 | -420.474 | -535.751 | 0.000 | 0.000 | 0.000 |
| -535.758 | -535.758 | -420.482 | 0.000 | -0.000 | -0.000 |
| -333.350 | -333.350 | -333.350 | 53.468 | -0.000 | -0.000 |
| -333.350 | -333.350 | -333.350 | 0.000 | 53.468 | -0.000 |
| -333.350 | -333.350 | -333.350 | -0.000 | 0.000 | 53.468 |
| 567.389 | 441.326 | 441.326 | 0.000 | 0.000 | 0.000 |
| 441.326 | 567.389 | 441.326 | -0.000 | -0.000 | -0.000 |
| 441.326 | 441.326 | 567.389 | 0.000 | 0.000 | -0.000 |
| 333.350 | 333.350 | 333.350 | 53.468 | 0.000 | 0.000 |
| 333.350 | 333.350 | 333.350 | 0.000 | 53.468 | -0.000 |
| 333.350 | 333.350 | 333.350 | -0.000 | 0.000 | 53.468 |
| 252.018 | 105.932 | 105.932 | 0.000 | -0.000 | -0.000 |
| 105.932 | 252.018 | 105.932 | -0.000 | -0.000 | 0.000 |
| 105.932 | 105.932 | 252.018 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 63.999 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 63.999 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 63.999 |
| 291.440 | 176.140 | 176.139 | 0.000 | 0.000 | -0.000 |
| 176.139 | 291.440 | 176.140 | 0.000 | 0.000 | 0.000 |
| 199.240 | 199.240 | 324.501 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 63.999 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 63.999 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 63.999 |
| 221.323 | 101.820 | 101.820 | -0.000 | -0.000 | 0.000 |
| 101.820 | 221.323 | 101.820 | -0.000 | -0.000 | -0.000 |
| 101.820 | 101.820 | 221.323 | 0.000 | -0.000 | -0.000 |
| -3.334 | -3.334 | -3.334 | 53.466 | -0.000 | -0.000 |
| -3.334 | -3.334 | -3.334 | 0.000 | 53.466 | -0.000 |
| -3.334 | -3.334 | -3.334 | -0.000 | -0.000 | 53.466 |
| 252.017 | 105.931 | 105.931 | -0.000 | 0.000 | -0.000 |
| 105.931 | 252.017 | 105.931 | -0.000 | 0.000 | 0.000 |
| 105.931 | 105.931 | 252.017 | -0.000 | 0.000 | -0.000 |
| 3.334 | 3.334 | 3.334 | 53.466 | 0.000 | 0.000 |
| 3.334 | 3.334 | 3.334 | -0.000 | 53.466 | 0.000 |
| 3.334 | 3.334 | 3.334 | 0.000 | 0.000 | 53.466 |
| 248.921 | 105.167 | 105.167 | 0.000 | 0.000 | -0.000 |
| 105.167 | 248.921 | 105.167 | 0.000 | 0.000 | -0.000 |
| 105.167 | 105.167 | 248.921 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 62.693 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 62.693 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 62.693 |
| -151.777 | -276.770 | -276.770 | -0.000 | -0.000 | 0.000 |
| -276.770 | -151.777 | -276.770 | -0.000 | -0.000 | 0.000 |
| -276.770 | -276.770 | -151.777 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 62.693 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 62.693 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 62.693 |
| 245.570 | 128.713 | 128.713 | 0.000 | 0.000 | -0.000 |
| 128.713 | 245.570 | 128.713 | -0.000 | -0.000 | -0.000 |
| 128.713 | 128.713 | 245.570 | -0.000 | -0.000 | -0.000 |
| 38.457 | 38.457 | 38.457 | 52.660 | 0.000 | 0.000 |
| 38.457 | 38.457 | 38.457 | 0.000 | 52.660 | -0.000 |
| 38.457 | 38.457 | 38.457 | 0.000 | 0.000 | 52.660 |
| 248.921 | 105.167 | 105.167 | -0.000 | -0.000 | 0.000 |
| 88.111 | 205.100 | 88.111 | 0.000 | 0.000 | 0.000 |
| 88.110 | 88.110 | 205.099 | 0.000 | 0.000 | 0.000 |
| -38.457 | -38.457 | -38.457 | 52.660 | -0.000 | -0.000 |
| -38.457 | -38.457 | -38.457 | 0.000 | 52.660 | -0.000 |
| -38.457 | -38.457 | -38.457 | -0.000 | -0.000 | 52.660 |
| 248.923 | 105.168 | 105.168 | 0.000 | 0.000 | -0.000 |
| 105.168 | 248.923 | 105.168 | 0.000 | 0.000 | -0.000 |
| 105.168 | 105.168 | 248.923 | -0.000 | 0.000 | 0.000 |
| 1.857 | 1.857 | 1.857 | 42.653 | -0.000 | 0.000 |
| 1.857 | 1.857 | 1.857 | -0.000 | 42.653 | -0.000 |
| 1.857 | 1.857 | 1.857 | -0.000 | -0.000 | 42.653 |
| 223.227 | 106.172 | 106.172 | 0.000 | 0.000 | -0.000 |
| 106.172 | 223.227 | 106.172 | 0.000 | -0.000 | -0.000 |
| 106.172 | 106.172 | 223.227 | 0.000 | 0.000 | 0.000 |
| -1.857 | -1.857 | -1.857 | 42.653 | 0.000 | -0.000 |
| -1.857 | -1.857 | -1.857 | 0.000 | 42.653 | -0.000 |
| -1.857 | -1.857 | -1.857 | 0.000 | -0.000 | 42.653 |
| 601.596 | 529.706 | 1554.669 | 0.000 | -0.000 | -1.568 |
| 529.706 | 601.596 | 1554.669 | -0.000 | -0.000 | 1.568 |
| 78.301 | 78.301 | 162.369 | -0.000 | -0.000 | -0.000 |
| 459.353 | 459.353 | 1479.000 | 25.850 | -0.000 | -0.000 |
| 459.353 | 459.353 | 1479.000 | 0.000 | 25.850 | -0.000 |
| 457.781 | 460.924 | 1478.998 | 0.000 | 0.000 | 12.506 |
| -317.186 | -389.002 | -1403.333 | 0.000 | 0.000 | -1.575 |
| -389.002 | -317.186 | -1403.333 | 0.000 | 0.000 | 1.575 |
| -379.869 | -379.869 | -1332.849 | -0.000 | 0.000 | 0.000 |
| -459.353 | -459.353 | -1479.000 | 25.850 | -0.000 | 0.000 |
| -459.353 | -459.353 | -1479.000 | 0.000 | 25.850 | 0.000 |
| -460.924 | -457.781 | -1478.998 | -0.000 | 0.000 | 12.506 |
| 158.920 | 71.262 | 78.301 | 0.000 | 0.000 | -0.130 |
| 71.262 | 158.920 | 78.301 | 0.000 | -0.000 | 0.130 |
| 872.170 | 872.170 | 560.879 | 0.000 | -0.000 | -0.000 |
| 45.930 | 45.930 | 147.881 | 25.851 | -0.000 | -0.000 |
| 45.930 | 45.930 | 147.881 | 0.000 | 25.851 | 0.000 |
| -0.130 | 0.130 | 0.000 | 0.000 | -0.000 | 28.788 |
| 95.901 | 24.418 | -72.222 | -0.000 | 0.000 | -1.605 |
| 24.418 | 95.901 | -72.222 | -0.000 | 0.000 | 1.605 |
| 78.301 | 78.301 | 162.369 | -0.000 | -0.000 | 0.000 |
| -45.930 | -45.930 | -147.881 | 25.851 | -0.000 | 0.000 |
| -45.930 | -45.930 | -147.881 | 0.000 | 25.851 | -0.000 |
| -0.130 | 0.130 | -0.000 | -0.000 | -0.000 | 28.788 |
| 150.139 | 75.040 | 90.699 | 0.000 | 0.000 | -1.275 |
| 75.040 | 150.139 | 90.699 | -0.000 | -0.000 | 1.275 |
| 117.567 | 117.567 | 157.478 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 35.712 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 35.712 | 0.000 |
| -0.130 | 0.130 | 0.000 | 0.000 | -0.000 | 28.788 |
| 134.355 | 65.880 | 61.335 | 0.000 | 0.000 | -1.880 |
| 65.880 | 134.355 | 61.335 | 0.000 | 0.000 | 1.880 |
| 74.859 | 74.859 | 133.047 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 35.712 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 35.712 | -0.000 |
| -0.130 | 0.130 | -0.000 | -0.000 | 0.000 | 28.788 |
| 139.439 | 70.271 | 78.515 | -0.000 | 0.000 | -0.000 |
| 70.271 | 139.439 | 78.515 | 0.000 | 0.000 | 0.000 |
| 78.046 | 78.046 | 152.445 | 0.000 | -0.000 | -0.000 |
| -1435.799 | -1435.799 | 173.606 | 27.332 | -0.000 | 0.000 |
| -1435.799 | -1435.799 | 173.606 | -0.000 | 27.332 | 0.000 |
| -1436.880 | -1434.719 | 173.606 | -0.000 | -0.000 | 15.564 |
| 1562.340 | 1504.465 | -93.838 | 0.000 | 0.000 | -1.080 |
| 1504.465 | 1562.340 | -93.838 | 0.000 | 0.000 | 1.080 |
| 1514.453 | 1514.453 | -37.022 | 0.000 | 0.000 | -0.000 |
| 1435.799 | 1435.799 | -173.606 | 27.332 | 0.000 | -0.000 |
| 1435.799 | 1435.799 | -173.606 | 0.000 | 27.332 | -0.000 |
| 1434.719 | 1436.880 | -173.606 | 0.000 | 0.000 | 15.564 |
| 139.439 | 70.271 | 78.515 | -0.000 | -0.000 | -0.000 |
| 70.271 | 139.439 | 78.515 | 0.000 | 0.000 | 0.000 |
| -64.910 | -64.910 | 153.872 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 35.320 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 35.320 | 0.000 |
| -144.671 | -142.510 | 17.362 | -0.000 | -0.000 | 15.563 |
| 139.439 | 70.271 | 78.515 | 0.000 | -0.000 | -0.000 |
| 70.271 | 139.439 | 78.515 | -0.000 | 0.000 | 0.000 |
| 222.223 | 222.223 | 119.283 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 35.320 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 35.320 | -0.000 |
| 142.510 | 144.671 | -17.362 | 0.000 | 0.000 | 15.563 |
| 111.868 | 54.310 | 81.533 | -0.000 | -0.000 | -1.111 |
| 54.310 | 111.868 | 81.533 | -0.000 | -0.000 | 1.111 |
| 78.046 | 78.046 | 152.446 | 0.000 | -0.000 | 0.000 |
| -32.150 | -32.150 | -5.145 | 19.824 | -0.000 | -0.000 |
| -32.150 | -32.150 | -5.145 | 0.000 | 19.824 | 0.000 |
| -15.543 | -13.367 | 1.748 | -0.000 | -0.000 | 15.476 |
| 139.439 | 70.271 | 78.515 | -0.000 | 0.000 | -0.000 |
| 70.271 | 139.439 | 78.515 | -0.000 | 0.000 | 0.000 |
| 138.761 | 138.761 | 120.095 | 0.000 | 0.000 | -0.000 |
| 32.150 | 32.150 | 5.145 | 19.824 | -0.000 | 0.000 |
| 32.150 | 32.150 | 5.145 | 0.000 | 19.824 | -0.000 |
| 13.367 | 15.543 | -1.748 | 0.000 | 0.000 | 15.476 |
| -259.192 | -957.605 | -1149.633 | 0.000 | 0.000 | 0.000 |
| 146.977 | 315.114 | 129.821 | 0.000 | -0.000 | 0.000 |
| 519.465 | -1242.512 | -1900.253 | 0.000 | -0.000 | 0.000 |
| -504.179 | -1085.157 | -1283.543 | 60.025 | 0.000 | 0.000 |
| 395.652 | -1355.202 | -2174.244 | 0.000 | 58.700 | 0.000 |
| 575.013 | -6011.770 | 1165.886 | 0.000 | 0.000 | 60.167 |
| -1360.048 | 2329.142 | 1271.945 | -0.000 | 0.000 | 0.000 |
| 631.529 | 1365.845 | 1404.152 | -0.000 | -0.000 | -0.000 |
| -271.874 | 1467.829 | 2448.156 | -0.000 | 0.000 | -0.000 |
| 504.179 | 1085.157 | 1283.543 | 60.025 | 0.000 | -0.000 |
| -395.601 | 1355.311 | 2174.374 | -0.000 | 58.699 | -0.000 |
| -1572.812 | 2196.064 | 1148.290 | -0.000 | -0.000 | 60.725 |
| 239.700 | -18.512 | -93.193 | 0.000 | 0.000 | 0.000 |
| 156.390 | 116.563 | -105.094 | 0.000 | 0.000 | -0.000 |
| 179.159 | -69.738 | 110.281 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 71.221 | -0.000 | 0.000 |
| 54.589 | -183.538 | -161.106 | 0.000 | 56.900 | 0.000 |
| 7.469 | -543.904 | 51.414 | 0.000 | 0.000 | 60.890 |
| 160.271 | 252.386 | 341.471 | -0.000 | 0.000 | -0.000 |
| 146.978 | 315.114 | 129.821 | -0.000 | -0.000 | -0.000 |
| 144.618 | 129.820 | 306.357 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 71.221 | 0.000 | -0.000 |
| 50.419 | 108.518 | 128.357 | -0.000 | 71.092 | -0.000 |
| -7.469 | 543.904 | -51.414 | -0.000 | -0.000 | 60.890 |
| 225.118 | 109.465 | 111.516 | 0.000 | 0.000 | 0.000 |
| 122.454 | 270.090 | 107.820 | 0.000 | -0.000 | 0.000 |
| 128.556 | 109.605 | 276.929 | 0.000 | 0.000 | 0.000 |
| 3.994 | -13.519 | -21.716 | 50.099 | 0.000 | 0.000 |
| 3.972 | -13.504 | -21.682 | 0.000 | 58.762 | 0.000 |
| -5.048 | -10.865 | -12.852 | 0.000 | -0.000 | 73.279 |
| 298.311 | 146.978 | 144.619 | -0.000 | -0.000 | -0.000 |
| 125.598 | 308.014 | 106.748 | -0.000 | -0.000 | -0.000 |
| 108.169 | 134.405 | 280.018 | -0.000 | -0.000 | 0.000 |
| -3.994 | 13.519 | 21.716 | 50.099 | 0.000 | -0.000 |
| -3.972 | 13.504 | 21.682 | -0.000 | 58.762 | -0.000 |
| 5.048 | 10.865 | 12.852 | -0.000 | -0.000 | 73.279 |
| 2152.090 | -5493.061 | 461.331 | -0.000 | 0.000 | -0.000 |
| 573.900 | -3306.412 | 332.387 | 0.000 | -0.000 | 0.000 |
| 568.963 | -3440.262 | 504.199 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 80.900 | -0.000 | 0.000 |
| 443.056 | -3566.439 | 204.619 | 0.000 | 58.269 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 96.799 |
| -252.910 | 3697.612 | -77.962 | -0.000 | -0.000 | 0.000 |
| -312.201 | 3826.472 | -76.842 | -0.000 | 0.000 | 0.000 |
| 152.342 | 145.987 | 339.443 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 80.900 | 0.000 | -0.000 |
| -443.056 | 3566.439 | -204.619 | -0.000 | 58.269 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 96.799 |
| 318.429 | -303.961 | 137.378 | -0.000 | 0.000 | -0.000 |
| 160.068 | 325.506 | 147.071 | 0.000 | -0.000 | -0.000 |
| 125.002 | -102.621 | 254.183 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 80.900 | 0.000 | 0.000 |
| -12.134 | -239.290 | -64.444 | -0.000 | 72.548 | 0.000 |
| 118.571 | -445.682 | 10.263 | -0.000 | 0.000 | 74.655 |
| 145.671 | 487.645 | 106.156 | -0.000 | -0.000 | 0.000 |
| 86.606 | 616.710 | 107.356 | -0.000 | 0.000 | 0.000 |
| 152.342 | 145.987 | 339.443 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 80.900 | -0.000 | -0.000 |
| 12.134 | 239.290 | 64.444 | 0.000 | 72.548 | -0.000 |
| -118.571 | 445.682 | -10.263 | 0.000 | -0.000 | 74.655 |
| 296.553 | 159.816 | 153.173 | 0.000 | -0.000 | -0.000 |
| 166.743 | 211.905 | 129.155 | 0.000 | -0.000 | 0.000 |
| 130.488 | 91.056 | 301.990 | 0.000 | -0.000 | 0.000 |
| -1.220 | -24.063 | -6.480 | 66.459 | 0.000 | 0.000 |
| 4.443 | -35.590 | 2.082 | 0.000 | 58.300 | -0.000 |
| 4.395 | -35.402 | 2.026 | 0.000 | -0.000 | 73.843 |
| 238.358 | 163.773 | 144.155 | 0.000 | -0.000 | -0.000 |
| 140.925 | 313.040 | 143.383 | 0.000 | -0.000 | -0.000 |
| 137.781 | 160.321 | 324.307 | 0.000 | -0.000 | -0.000 |
| 1.220 | 24.063 | 6.480 | 66.459 | -0.000 | -0.000 |
| -4.443 | 35.590 | -2.082 | -0.000 | 58.300 | 0.000 |
| -4.395 | 35.402 | -2.026 | -0.000 | 0.000 | 73.843 |
| 224.419 | 117.771 | 117.771 | 0.000 | 0.000 | 0.000 |
| 117.771 | 224.419 | 117.771 | 0.000 | 0.000 | -0.000 |
| 117.771 | 117.771 | 224.419 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 94.951 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 94.951 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 94.951 |
| 224.419 | 117.771 | 117.771 | -0.000 | 0.000 | -0.000 |
| 117.771 | 224.419 | 117.771 | -0.000 | -0.000 | 0.000 |
| 117.771 | 117.771 | 224.419 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 94.951 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 94.951 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 94.951 |
| 224.419 | 117.771 | 117.771 | -0.000 | 0.000 | 0.000 |
| 117.771 | 224.419 | 117.771 | -0.000 | 0.000 | -0.000 |
| 117.771 | 117.771 | 224.419 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 94.951 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 94.951 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 94.951 |
| 224.419 | 117.771 | 117.771 | -0.000 | -0.000 | -0.000 |
| 117.771 | 224.419 | 117.771 | -0.000 | -0.000 | -0.000 |
| 117.771 | 117.771 | 224.419 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 94.951 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 94.951 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 94.951 |
| 224.420 | 117.771 | 117.771 | 0.000 | 0.000 | 0.000 |
| 117.771 | 224.420 | 117.771 | -0.000 | 0.000 | -0.000 |
| 117.771 | 117.771 | 224.420 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 94.951 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 94.951 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 94.951 |
| 224.419 | 117.770 | 117.770 | -0.000 | -0.000 | -0.000 |
| 117.770 | 224.419 | 117.770 | -0.000 | -0.000 | -0.000 |
| 117.770 | 117.770 | 224.419 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 94.951 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 94.951 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 94.951 |
| 1.798 | 10.415 | 10.415 | -0.000 | -0.000 | -0.000 |
| 10.415 | 1.798 | 10.415 | 0.000 | -0.000 | -0.000 |
| 10.415 | 10.415 | 1.798 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 10.013 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 10.013 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 10.013 |
| 1.798 | 10.415 | 10.415 | -0.000 | -0.000 | -0.000 |
| 10.415 | 1.798 | 10.415 | -0.000 | -0.000 | 0.000 |
| 10.415 | 10.415 | 1.798 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 10.013 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 10.013 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 10.013 |
| 1.798 | 10.415 | 10.415 | -0.000 | 0.000 | 0.000 |
| 10.415 | 1.798 | 10.415 | -0.000 | -0.000 | -0.000 |
| 10.415 | 10.415 | 1.798 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -2.588 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -2.588 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | -2.588 |
| 1.798 | 10.415 | 10.415 | 0.000 | 0.000 | 0.000 |
| 10.415 | 1.798 | 10.415 | 0.000 | -0.000 | 0.000 |
| 10.415 | 10.415 | 1.798 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -2.588 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -2.588 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -2.588 |
| 1.798 | 10.415 | 10.415 | 0.000 | 0.000 | -0.000 |
| 10.415 | 1.798 | 10.415 | -0.000 | 0.000 | -0.000 |
| 10.415 | 10.415 | 1.798 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -2.588 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -2.588 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -2.588 |
| 1.798 | 10.415 | 10.415 | -0.000 | -0.000 | -0.000 |
| 10.415 | 1.798 | 10.415 | -0.000 | -0.000 | 0.000 |
| 10.415 | 10.415 | 1.798 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -2.588 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -2.588 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -2.588 |
| 581.520 | 302.017 | 302.018 | 0.000 | -0.000 | 0.000 |
| 302.017 | 581.520 | 302.018 | -0.000 | 0.000 | 0.000 |
| 302.017 | 302.017 | 581.520 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 207.415 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 207.415 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 207.415 |
| 581.520 | 302.018 | 302.018 | 0.000 | 0.000 | 0.000 |
| 302.018 | 581.520 | 302.018 | 0.000 | -0.000 | -0.000 |
| 302.018 | 302.018 | 581.520 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 207.415 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 207.415 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 207.415 |
| 581.520 | 302.017 | 302.017 | -0.000 | 0.000 | 0.000 |
| 302.017 | 581.520 | 302.017 | 0.000 | 0.000 | 0.000 |
| 302.017 | 302.017 | 581.520 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 207.415 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 207.415 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 207.415 |
| 581.521 | 302.018 | 302.018 | 0.000 | -0.000 | -0.000 |
| 302.018 | 581.521 | 302.018 | -0.000 | 0.000 | -0.000 |
| 302.018 | 302.018 | 581.521 | 0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 207.415 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 207.415 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 207.415 |
| 581.515 | 302.011 | 302.011 | -0.000 | 0.000 | 0.000 |
| 302.011 | 581.515 | 302.011 | 0.000 | -0.000 | 0.000 |
| 302.011 | 302.011 | 581.515 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 207.415 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 207.415 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 207.415 |
| 581.526 | 302.024 | 302.024 | 0.000 | 0.000 | 0.000 |
| 302.024 | 581.526 | 302.024 | 0.000 | 0.000 | -0.000 |
| 302.024 | 302.024 | 581.526 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 207.415 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 207.415 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 207.415 |
| 25.668 | 2.278 | 2.278 | 0.000 | 0.000 | 0.000 |
| 2.278 | 25.668 | 2.278 | 0.000 | 0.000 | -0.000 |
| 2.278 | 2.278 | 25.668 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.434 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -0.434 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -0.434 |
| 25.668 | 2.278 | 2.278 | -0.000 | -0.000 | -0.000 |
| 2.278 | 25.668 | 2.278 | -0.000 | -0.000 | -0.000 |
| 2.278 | 2.278 | 25.668 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.434 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.434 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -0.434 |
| 25.668 | 2.278 | 2.278 | 0.000 | 0.000 | 0.000 |
| 2.278 | 25.668 | 2.278 | 0.000 | 0.000 | 0.000 |
| 2.278 | 2.278 | 25.668 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.434 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -0.434 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | -0.434 |
| 25.668 | 2.278 | 2.278 | -0.000 | -0.000 | -0.000 |
| 2.278 | 25.668 | 2.278 | -0.000 | -0.000 | -0.000 |
| 2.278 | 2.278 | 25.668 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.434 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.434 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -0.434 |
| 25.668 | 2.279 | 2.279 | 0.000 | 0.000 | 0.000 |
| 2.279 | 25.668 | 2.279 | 0.000 | 0.000 | 0.000 |
| 2.279 | 2.279 | 25.668 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.434 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.434 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | -0.434 |
| 25.668 | 2.278 | 2.278 | -0.000 | -0.000 | -0.000 |
| 2.278 | 25.668 | 2.278 | -0.000 | -0.000 | -0.000 |
| 2.278 | 2.278 | 25.668 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.434 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.434 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -0.434 |
| 147.981 | 107.908 | 107.908 | -0.000 | 0.000 | 0.000 |
| 107.908 | 147.981 | 107.908 | 0.000 | 0.000 | 0.000 |
| 107.908 | 107.908 | 147.981 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 32.809 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 32.809 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 32.809 |
| 147.981 | 107.908 | 107.908 | -0.000 | -0.000 | -0.000 |
| 107.908 | 147.981 | 107.908 | 0.000 | -0.000 | -0.000 |
| 107.908 | 107.908 | 147.981 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 32.809 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 32.809 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 32.809 |
| 147.981 | 107.908 | 107.908 | 0.000 | 0.000 | 0.000 |
| 107.908 | 147.981 | 107.908 | 0.000 | 0.000 | 0.000 |
| 107.908 | 107.908 | 147.981 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 75.196 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 75.196 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 75.196 |
| 147.981 | 107.908 | 107.908 | -0.000 | -0.000 | -0.000 |
| 107.908 | 147.981 | 107.908 | 0.000 | -0.000 | -0.000 |
| 107.908 | 107.908 | 147.981 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 75.196 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 75.196 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 75.196 |
| 147.979 | 107.906 | 107.906 | 0.000 | 0.000 | 0.000 |
| 107.906 | 147.979 | 107.906 | 0.000 | 0.000 | 0.000 |
| 107.906 | 107.906 | 147.979 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 32.809 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 32.809 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 32.809 |
| 147.982 | 107.910 | 107.910 | -0.000 | -0.000 | -0.000 |
| 107.910 | 147.982 | 107.910 | -0.000 | -0.000 | -0.000 |
| 107.910 | 107.910 | 147.982 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 32.809 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 32.809 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 32.809 |
| 366.815 | 116.705 | 116.705 | 0.000 | 0.000 | 0.000 |
| 116.705 | 366.815 | 116.705 | -0.000 | -0.000 | 0.000 |
| 116.705 | 116.705 | 366.815 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 76.087 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 76.087 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 76.087 |
| 366.815 | 116.705 | 116.705 | 0.000 | 0.000 | 0.000 |
| 116.705 | 366.815 | 116.705 | -0.000 | 0.000 | -0.000 |
| 116.705 | 116.705 | 366.815 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 76.087 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 76.087 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 76.087 |
| 366.816 | 116.705 | 116.705 | 0.000 | -0.000 | 0.000 |
| 116.705 | 366.816 | 116.705 | -0.000 | -0.000 | 0.000 |
| 116.705 | 116.705 | 366.816 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 76.087 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 76.087 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 76.087 |
| 366.804 | 116.710 | 116.710 | 0.000 | 0.000 | -0.000 |
| 116.710 | 366.804 | 116.710 | -0.000 | -0.000 | -0.000 |
| 116.710 | 116.710 | 366.804 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 76.087 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 76.087 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 76.087 |
| 366.806 | 116.711 | 116.711 | 0.000 | -0.000 | -0.000 |
| 116.711 | 366.806 | 116.711 | 0.000 | 0.000 | 0.000 |
| 116.711 | 116.711 | 366.806 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 76.087 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 76.087 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 76.087 |
| 366.803 | 116.710 | 116.710 | 0.000 | 0.000 | -0.000 |
| 116.710 | 366.803 | 116.710 | -0.000 | 0.000 | -0.000 |
| 116.710 | 116.710 | 366.803 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 76.087 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 76.087 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 76.087 |
| 14.036 | 3.339 | 3.339 | 0.000 | 0.000 | -0.000 |
| 3.339 | 14.036 | 3.339 | 0.000 | 0.000 | -0.000 |
| 3.339 | 3.339 | 14.036 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 2.928 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 2.928 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 2.928 |
| 13.673 | 3.521 | 3.521 | 0.000 | 0.000 | 0.000 |
| 3.521 | 13.673 | 3.521 | 0.000 | -0.000 | 0.000 |
| 3.521 | 3.521 | 13.673 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 2.928 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 2.928 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 2.928 |
| 13.673 | 3.521 | 3.521 | -0.000 | 0.000 | -0.000 |
| 3.521 | 13.673 | 3.521 | -0.000 | 0.000 | -0.000 |
| 3.521 | 3.521 | 13.673 | -0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 2.928 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 2.928 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 2.928 |
| 14.036 | 3.339 | 3.339 | 0.000 | -0.000 | 0.000 |
| 3.339 | 14.036 | 3.339 | 0.000 | -0.000 | 0.000 |
| 3.339 | 3.339 | 14.036 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 2.928 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 2.928 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 2.928 |
| 14.036 | 3.339 | 3.339 | -0.000 | 0.000 | -0.000 |
| 3.339 | 14.036 | 3.339 | -0.000 | 0.000 | -0.000 |
| 3.339 | 3.339 | 14.036 | -0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 3.180 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 3.180 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 3.180 |
| 14.036 | 3.339 | 3.339 | 0.000 | -0.000 | 0.000 |
| 3.339 | 14.036 | 3.339 | 0.000 | -0.000 | 0.000 |
| 3.339 | 3.339 | 14.036 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 3.180 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 3.180 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 3.180 |
| 306.231 | 239.260 | 239.260 | -0.000 | -0.000 | -0.000 |
| 239.260 | 306.231 | 239.260 | -0.000 | -0.000 | -0.000 |
| 239.260 | 239.260 | 306.231 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 177.826 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 177.826 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 177.826 |
| 306.231 | 239.260 | 239.260 | 0.000 | 0.000 | 0.000 |
| 239.260 | 306.231 | 239.260 | 0.000 | -0.000 | 0.000 |
| 239.260 | 239.260 | 306.231 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 177.826 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 177.826 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 177.826 |
| 306.231 | 239.260 | 239.260 | -0.000 | -0.000 | -0.000 |
| 239.260 | 306.231 | 239.260 | -0.000 | 0.000 | -0.000 |
| 239.260 | 239.260 | 306.231 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 190.364 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 190.364 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 190.364 |
| 306.231 | 239.260 | 239.260 | 0.000 | -0.000 | 0.000 |
| 239.260 | 306.231 | 239.260 | 0.000 | 0.000 | -0.000 |
| 239.260 | 239.260 | 306.231 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 190.364 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 190.364 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 190.364 |
| 306.232 | 239.259 | 239.259 | -0.000 | -0.000 | -0.000 |
| 239.259 | 306.232 | 239.259 | -0.000 | -0.000 | -0.000 |
| 239.259 | 239.259 | 306.232 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.001 | 177.826 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.001 | -0.000 | 177.826 | -0.000 |
| 0.000 | 0.001 | 0.000 | -0.000 | 0.000 | 177.826 |
| 306.229 | 239.262 | 239.262 | 0.000 | -0.000 | 0.000 |
| 239.262 | 306.229 | 239.262 | 0.000 | 0.000 | -0.000 |
| 239.262 | 239.262 | 306.229 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.001 | 177.826 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.001 | 0.000 | 177.826 | -0.000 |
| -0.000 | -0.001 | -0.000 | 0.000 | -0.000 | 177.826 |
| 208.345 | 173.844 | 173.844 | -0.000 | 0.000 | 0.000 |
| 173.844 | 208.345 | 173.844 | 0.000 | 0.000 | 0.000 |
| 173.844 | 173.844 | 208.345 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 101.657 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 101.657 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 101.657 |
| 208.345 | 173.844 | 173.844 | -0.000 | -0.000 | 0.000 |
| 173.844 | 208.345 | 173.844 | -0.000 | -0.000 | 0.000 |
| 173.844 | 173.844 | 208.345 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 101.657 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 101.657 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 101.657 |
| 208.345 | 173.845 | 173.845 | 0.000 | 0.000 | -0.000 |
| 173.845 | 208.345 | 173.845 | 0.000 | 0.000 | -0.000 |
| 173.845 | 173.845 | 208.345 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 101.657 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 101.657 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 101.657 |
| 208.345 | 173.844 | 173.844 | -0.000 | 0.000 | -0.000 |
| 173.844 | 208.345 | 173.844 | 0.000 | -0.000 | 0.000 |
| 173.844 | 173.844 | 208.345 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 101.657 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 101.657 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 101.657 |
| 208.348 | 173.848 | 173.848 | -0.000 | 0.000 | 0.000 |
| 173.848 | 208.348 | 173.848 | 0.000 | -0.000 | -0.000 |
| 173.848 | 173.848 | 208.348 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 101.657 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 101.657 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 101.657 |
| 208.342 | 173.841 | 173.841 | 0.000 | -0.000 | -0.000 |
| 173.841 | 208.342 | 173.841 | -0.000 | 0.000 | -0.000 |
| 173.841 | 173.841 | 208.342 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 101.657 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 101.657 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 101.657 |
| 172.043 | 184.286 | 142.626 | 0.000 | 0.000 | -0.000 |
| 184.286 | 172.043 | 142.626 | 0.000 | 0.000 | -0.000 |
| 142.626 | 142.626 | 219.303 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 76.478 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 76.478 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 122.401 |
| 172.043 | 184.286 | 142.626 | 0.000 | 0.000 | 0.000 |
| 184.286 | 172.043 | 142.626 | -0.000 | 0.000 | 0.000 |
| 142.626 | 142.626 | 219.303 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 76.478 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 76.478 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 122.401 |
| 172.043 | 184.286 | 142.626 | -0.000 | -0.000 | -0.000 |
| 184.286 | 172.043 | 142.626 | -0.000 | -0.000 | 0.000 |
| 142.625 | 142.625 | 219.303 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 87.166 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 87.166 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 122.401 |
| 142.879 | 213.449 | 142.625 | -0.000 | 0.000 | 0.000 |
| 213.449 | 142.879 | 142.625 | 0.000 | -0.000 | 0.000 |
| 142.626 | 142.626 | 219.303 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 87.166 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 87.166 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 122.401 |
| 142.880 | 213.450 | 142.626 | -0.000 | 0.000 | -0.000 |
| 213.450 | 142.880 | 142.626 | -0.000 | 0.000 | -0.000 |
| 142.625 | 142.625 | 219.300 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 76.478 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 76.478 | -0.000 |
| 0.000 | 0.001 | 0.000 | -0.000 | -0.000 | 122.401 |
| 142.879 | 213.448 | 142.625 | 0.000 | -0.000 | -0.000 |
| 213.448 | 142.879 | 142.625 | 0.000 | -0.000 | 0.000 |
| 142.626 | 142.626 | 219.305 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 76.478 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 76.478 | 0.000 |
| -0.000 | -0.001 | -0.000 | 0.000 | -0.000 | 122.401 |
| 410.783 | 265.121 | 265.121 | -0.000 | 0.000 | -0.000 |
| 265.121 | 410.783 | 265.121 | 0.000 | 0.000 | -0.000 |
| 265.121 | 265.121 | 410.783 | -0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 117.574 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 117.574 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 117.574 |
| -727.125 | -858.138 | -858.138 | 0.000 | 0.000 | -0.000 |
| -858.138 | -727.125 | -858.138 | 0.000 | 0.000 | -0.000 |
| -858.138 | -858.138 | -727.125 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 117.574 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 117.574 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 117.574 |
| 496.976 | 383.207 | 383.207 | -0.000 | 0.000 | -0.000 |
| 383.207 | 496.976 | 383.207 | -0.000 | 0.000 | 0.000 |
| 265.120 | 265.120 | 410.782 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 117.574 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 117.574 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 117.574 |
| 410.784 | 265.122 | 265.122 | 0.000 | -0.000 | 0.000 |
| 265.122 | 410.784 | 265.122 | -0.000 | -0.000 | 0.000 |
| 265.122 | 265.122 | 410.784 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 117.574 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 117.574 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 117.574 |
| 410.773 | 265.111 | 265.111 | -0.000 | 0.000 | -0.000 |
| 265.111 | 410.773 | 265.111 | 0.000 | 0.000 | -0.000 |
| 280.220 | 280.220 | 407.742 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 117.574 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 117.574 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 117.574 |
| 410.794 | 265.131 | 265.131 | 0.000 | -0.000 | 0.000 |
| 265.131 | 410.794 | 265.131 | -0.000 | -0.000 | 0.000 |
| 265.131 | 265.131 | 410.794 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 117.574 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 117.574 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 117.574 |
| 384.305 | 238.808 | 238.808 | 0.000 | 0.000 | -0.000 |
| 238.808 | 384.305 | 238.808 | 0.000 | 0.000 | 0.000 |
| 238.808 | 238.808 | 384.305 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 117.335 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 117.335 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 117.335 |
| 384.305 | 238.808 | 238.808 | -0.000 | -0.000 | 0.000 |
| 238.808 | 384.305 | 238.808 | -0.000 | -0.000 | 0.000 |
| 238.808 | 238.808 | 384.305 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 117.335 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 117.335 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 117.335 |
| 363.660 | 241.657 | 241.657 | -0.000 | 0.000 | -0.000 |
| 241.657 | 363.660 | 241.657 | 0.000 | -0.000 | -0.000 |
| 241.657 | 241.657 | 363.660 | 0.000 | 0.000 | -0.000 |
| 0.043 | 0.043 | 0.043 | 100.704 | 0.000 | -0.000 |
| 0.043 | 0.043 | 0.043 | 0.000 | 100.704 | -0.000 |
| 0.043 | 0.043 | 0.043 | 0.000 | -0.000 | 100.704 |
| 359.262 | 245.497 | 245.497 | -0.000 | 0.000 | 0.000 |
| 245.497 | 359.262 | 245.497 | -0.000 | 0.000 | 0.000 |
| 245.497 | 245.497 | 359.262 | -0.000 | 0.000 | 0.000 |
| -0.043 | -0.043 | -0.043 | 100.704 | 0.000 | 0.000 |
| -0.043 | -0.043 | -0.043 | -0.000 | 100.704 | 0.000 |
| -0.043 | -0.043 | -0.043 | -0.000 | -0.000 | 100.704 |
| 363.626 | 241.595 | 241.595 | 0.000 | 0.000 | -0.000 |
| 241.595 | 363.626 | 241.595 | 0.000 | -0.000 | -0.000 |
| 241.595 | 241.595 | 363.626 | 0.000 | 0.000 | -0.000 |
| 0.004 | 0.004 | 0.005 | 100.705 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 117.335 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 117.335 |
| 384.315 | 238.818 | 238.818 | -0.000 | -0.000 | 0.000 |
| 238.818 | 384.315 | 238.818 | -0.000 | -0.000 | 0.000 |
| 238.818 | 238.818 | 384.315 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 117.335 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 117.335 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 117.335 |
| 139.759 | 25.899 | 25.899 | 0.000 | 0.000 | -0.000 |
| 25.899 | 139.759 | 25.899 | 0.000 | -0.000 | -0.000 |
| 25.899 | 25.899 | 139.759 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 117.596 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 117.596 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 117.596 |
| 412.729 | 267.044 | 267.044 | 0.000 | -0.000 | -0.000 |
| 267.044 | 412.729 | 267.044 | 0.000 | -0.000 | -0.000 |
| 267.044 | 267.044 | 412.729 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 117.596 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 117.596 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 117.596 |
| 412.728 | 267.043 | 267.043 | -0.000 | 0.000 | -0.000 |
| 267.043 | 412.728 | 267.043 | -0.000 | 0.000 | 0.000 |
| 267.043 | 267.043 | 412.728 | -0.000 | 0.000 | -0.000 |
| -25.026 | -25.026 | -25.026 | 103.311 | 0.000 | -0.000 |
| -25.026 | -25.026 | -25.026 | 0.000 | 103.311 | -0.000 |
| -25.026 | -25.026 | -25.026 | 0.000 | -0.000 | 103.311 |
| 424.082 | 293.627 | 293.627 | -0.000 | -0.000 | 0.000 |
| 293.627 | 424.082 | 293.627 | 0.000 | -0.000 | 0.000 |
| 293.627 | 293.627 | 424.082 | -0.000 | -0.000 | 0.000 |
| 25.026 | 25.026 | 25.026 | 103.311 | -0.000 | 0.000 |
| 25.026 | 25.026 | 25.026 | -0.000 | 103.311 | 0.000 |
| 25.026 | 25.026 | 25.026 | 0.000 | 0.000 | 103.311 |
| 384.909 | 271.341 | 271.341 | -0.000 | -0.000 | -0.000 |
| 271.341 | 384.909 | 271.341 | 0.000 | 0.000 | -0.000 |
| 271.341 | 271.341 | 384.909 | 0.000 | -0.000 | 0.000 |
| -2.913 | -2.913 | -2.913 | 100.969 | 0.000 | -0.000 |
| -2.913 | -2.913 | -2.913 | 0.000 | 100.969 | -0.000 |
| -2.913 | -2.913 | -2.913 | 0.000 | -0.000 | 100.969 |
| 412.740 | 267.054 | 267.054 | 0.000 | -0.000 | -0.000 |
| 267.054 | 412.740 | 267.054 | 0.000 | -0.000 | 0.000 |
| 267.054 | 267.054 | 412.740 | 0.000 | -0.000 | 0.000 |
| 2.913 | 2.913 | 2.913 | 100.969 | 0.000 | 0.000 |
| 2.913 | 2.913 | 2.913 | -0.000 | 100.969 | 0.000 |
| 2.913 | 2.913 | 2.913 | -0.000 | 0.000 | 100.969 |
| 172.225 | 62.149 | 62.149 | 0.000 | 0.000 | -0.000 |
| 62.149 | 172.225 | 62.149 | 0.000 | -0.000 | 0.000 |
| 62.149 | 62.149 | 172.225 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 17.826 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 17.826 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 17.826 |
| 172.225 | 62.149 | 62.149 | -0.000 | 0.000 | -0.000 |
| 62.149 | 172.225 | 62.149 | -0.000 | -0.000 | -0.000 |
| 62.149 | 62.149 | 172.225 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 17.826 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 17.826 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 17.826 |
| 172.225 | 62.150 | 62.150 | 0.000 | -0.000 | -0.000 |
| 62.150 | 172.225 | 62.150 | 0.000 | -0.000 | 0.000 |
| 62.150 | 62.150 | 172.225 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 19.152 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 19.152 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 19.152 |
| 172.283 | 62.120 | 62.120 | -0.000 | -0.000 | -0.000 |
| 62.120 | 172.283 | 62.120 | -0.000 | 0.000 | 0.000 |
| 62.120 | 62.120 | 172.283 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 19.152 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 19.152 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 19.152 |
| 172.286 | 62.121 | 62.121 | 0.000 | -0.000 | 0.000 |
| 62.121 | 172.286 | 62.121 | 0.000 | -0.000 | -0.000 |
| 62.121 | 62.121 | 172.286 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 19.152 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 19.152 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 19.152 |
| 172.281 | 62.119 | 62.119 | -0.000 | 0.000 | -0.000 |
| 62.119 | 172.281 | 62.119 | -0.000 | 0.000 | 0.000 |
| 62.119 | 62.119 | 172.281 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 19.152 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 19.152 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 19.152 |
| 119.809 | 124.395 | 124.395 | 0.000 | 0.000 | 0.000 |
| 124.395 | 119.809 | 124.395 | 0.000 | 0.000 | -0.000 |
| 124.395 | 124.395 | 119.809 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 66.770 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 66.770 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 66.770 |
| 119.809 | 124.395 | 124.395 | -0.000 | -0.000 | -0.000 |
| 124.395 | 119.809 | 124.395 | -0.000 | -0.000 | -0.000 |
| 124.395 | 124.395 | 119.809 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 66.770 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 66.770 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 66.770 |
| 119.809 | 124.395 | 124.395 | 0.000 | 0.000 | 0.000 |
| 124.395 | 119.809 | 124.395 | 0.000 | 0.000 | 0.000 |
| 124.395 | 124.395 | 119.809 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 66.770 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 66.770 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 66.770 |
| 119.809 | 124.395 | 124.395 | -0.000 | -0.000 | -0.000 |
| 124.395 | 119.809 | 124.395 | -0.000 | -0.000 | -0.000 |
| 124.395 | 124.395 | 119.809 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 66.770 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 66.770 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 66.770 |
| 119.810 | 124.396 | 124.396 | 0.000 | 0.000 | 0.000 |
| 124.396 | 119.810 | 124.396 | 0.000 | 0.000 | -0.000 |
| 124.396 | 124.396 | 119.810 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 66.770 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 66.770 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 66.770 |
| 119.809 | 124.395 | 124.395 | -0.000 | -0.000 | -0.000 |
| 124.395 | 119.809 | 124.395 | -0.000 | -0.000 | -0.000 |
| 124.395 | 124.395 | 119.809 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 66.770 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 66.770 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 66.770 |
| 98.830 | 70.706 | 70.706 | -0.000 | -0.000 | 0.000 |
| 70.706 | 98.830 | 70.706 | -0.000 | -0.000 | 0.000 |
| 70.706 | 70.706 | 98.830 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 27.563 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 27.563 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 27.563 |
| 98.830 | 70.706 | 70.706 | 0.000 | -0.000 | -0.000 |
| 70.706 | 98.830 | 70.706 | -0.000 | -0.000 | 0.000 |
| 70.706 | 70.706 | 98.830 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 27.562 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 27.562 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 27.562 |
| 98.830 | 70.706 | 70.706 | 0.000 | 0.000 | -0.000 |
| 70.706 | 98.830 | 70.706 | 0.000 | 0.000 | 0.000 |
| 70.706 | 70.706 | 98.830 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 27.563 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 27.563 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 27.563 |
| 98.830 | 70.705 | 70.705 | 0.000 | -0.000 | -0.000 |
| 70.705 | 98.830 | 70.705 | -0.000 | -0.000 | -0.000 |
| 70.705 | 70.705 | 98.830 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 27.563 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 27.563 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 27.563 |
| 98.831 | 70.707 | 70.707 | 0.000 | -0.000 | 0.000 |
| 70.707 | 98.831 | 70.707 | -0.000 | 0.000 | 0.000 |
| 70.707 | 70.707 | 98.831 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 27.563 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 27.563 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 27.563 |
| 98.830 | 70.704 | 70.704 | 0.000 | -0.000 | -0.000 |
| 70.704 | 98.830 | 70.704 | -0.000 | 0.000 | -0.000 |
| 70.704 | 70.704 | 98.830 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 27.563 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 27.563 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 27.563 |
| 101.502 | 87.754 | 81.873 | -0.000 | 0.000 | 0.000 |
| 87.754 | 101.502 | 81.873 | 0.000 | -0.000 | 0.000 |
| 81.873 | 81.873 | 117.868 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 28.769 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 28.769 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 34.367 |
| 101.502 | 87.754 | 81.873 | -0.000 | -0.000 | -0.000 |
| 87.754 | 101.502 | 81.873 | -0.000 | -0.000 | -0.000 |
| 81.873 | 81.873 | 117.868 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 28.769 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 28.769 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 34.367 |
| 101.502 | 87.754 | 81.873 | 0.000 | 0.000 | 0.000 |
| 87.754 | 101.502 | 81.873 | 0.000 | 0.000 | 0.000 |
| 81.873 | 81.873 | 117.868 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 28.769 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 28.769 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 34.367 |
| 101.502 | 87.754 | 81.873 | -0.000 | -0.000 | -0.000 |
| 87.754 | 101.502 | 81.873 | -0.000 | -0.000 | -0.000 |
| 81.873 | 81.873 | 117.868 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 28.769 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 28.769 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 34.367 |
| 101.503 | 87.756 | 81.874 | 0.000 | 0.000 | 0.000 |
| 87.756 | 101.503 | 81.874 | 0.000 | 0.000 | 0.000 |
| 81.875 | 81.875 | 117.869 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 28.769 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 28.769 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 34.367 |
| 106.636 | 82.618 | 81.871 | -0.000 | -0.000 | 0.000 |
| 82.618 | 106.636 | 81.871 | -0.000 | -0.000 | -0.000 |
| 81.871 | 81.871 | 117.866 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 28.769 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 28.769 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 34.367 |
| 1008.612 | 925.616 | 457.219 | 11.209 | 0.000 | -0.000 |
| 923.103 | 1011.101 | 457.233 | -16.425 | 0.000 | 0.000 |
| 110.539 | 110.539 | 298.417 | -0.000 | 0.000 | -0.000 |
| 798.533 | 765.684 | 344.391 | 35.745 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 44.025 | 23.705 |
| 782.115 | 782.115 | 344.380 | -0.000 | 16.425 | 43.999 |
| -675.114 | -783.865 | 287.002 | 21.499 | -0.000 | 0.000 |
| 126.977 | 254.753 | 110.539 | -23.705 | -0.000 | -0.000 |
| -805.247 | -805.247 | 468.951 | 0.000 | -0.000 | 0.000 |
| -894.199 | -937.182 | 173.333 | 40.983 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 44.025 | 23.705 |
| -782.115 | -782.115 | -344.380 | 0.000 | 16.425 | 43.999 |
| 254.753 | 126.977 | 110.539 | 23.705 | 0.000 | -0.000 |
| 126.977 | 254.753 | 110.539 | -23.705 | 0.000 | -0.000 |
| 202.122 | 202.122 | 278.384 | -0.000 | 0.000 | -0.000 |
| 94.634 | 61.819 | 34.589 | 35.727 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 44.025 | 23.705 |
| 78.257 | 78.257 | 34.595 | -0.000 | 14.068 | 45.670 |
| 148.988 | 39.768 | 130.995 | 21.554 | 0.000 | 0.000 |
| 39.768 | 148.988 | 130.995 | -21.554 | -0.000 | 0.000 |
| 34.635 | 34.635 | 258.906 | 0.000 | -0.000 | 0.000 |
| 23.705 | -23.705 | -0.000 | 44.025 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 44.025 | 23.705 |
| -78.129 | -78.129 | -34.512 | 0.000 | 16.420 | 43.994 |
| 254.752 | 126.975 | 110.537 | 23.705 | 0.000 | -0.000 |
| 126.975 | 254.752 | 110.537 | -23.705 | 0.000 | -0.000 |
| 120.667 | 120.667 | 296.786 | -0.000 | 0.000 | 0.000 |
| 31.620 | -10.744 | -1.975 | 40.557 | 0.000 | -0.000 |
| 7.903 | 7.903 | 4.503 | -0.000 | 34.479 | 15.230 |
| 10.777 | 10.777 | -2.040 | -0.000 | 21.100 | 52.656 |
| 230.213 | 120.188 | 115.602 | 21.647 | -0.000 | -0.000 |
| 126.979 | 254.755 | 110.540 | -23.705 | -0.000 | -0.000 |
| 110.540 | 110.540 | 298.417 | 0.000 | -0.000 | 0.000 |
| 10.744 | -31.620 | 1.975 | 40.557 | -0.000 | 0.000 |
| -7.903 | -7.903 | -4.503 | 0.000 | 34.479 | 15.230 |
| -10.777 | -10.777 | 2.040 | 0.000 | 21.100 | 52.656 |
| 255.882 | 129.263 | 111.442 | -23.999 | 0.000 | 0.000 |
| 130.539 | 237.144 | 115.210 | 21.625 | 0.000 | -0.000 |
| 115.040 | 115.040 | 292.631 | 0.000 | -0.000 | -0.000 |
| -23.999 | 23.999 | -0.000 | 42.306 | -0.000 | 0.000 |
| -1.443 | -1.443 | 0.739 | 0.000 | 31.451 | -15.680 |
| -0.000 | 0.000 | -0.000 | 0.000 | -23.999 | 63.309 |
| 237.261 | 131.445 | 114.920 | -21.532 | -0.000 | 0.000 |
| 129.263 | 255.882 | 111.442 | 23.999 | -0.000 | -0.000 |
| 111.592 | 111.592 | 295.957 | -0.000 | -0.000 | -0.000 |
| -14.237 | 17.123 | -0.739 | 31.451 | -0.000 | 0.000 |
| 1.443 | 1.443 | -0.739 | -0.000 | 31.451 | -15.680 |
| 0.000 | 0.000 | 0.000 | 0.000 | -23.999 | 63.309 |
| 237.203 | 130.937 | 115.081 | -21.585 | 0.000 | -0.000 |
| 129.263 | 255.882 | 111.442 | 23.999 | 0.000 | -0.000 |
| 115.267 | 115.267 | 292.588 | 0.000 | 0.000 | -0.000 |
| -23.999 | 23.999 | 0.000 | 42.306 | 0.000 | -0.000 |
| 0.051 | 0.051 | -0.017 | 0.000 | 30.197 | -11.560 |
| 0.000 | 0.000 | 0.000 | 0.000 | -23.999 | 63.309 |
| 237.214 | 131.028 | 115.052 | -21.576 | -0.000 | -0.000 |
| 131.028 | 237.214 | 115.052 | 21.576 | 0.000 | 0.000 |
| 113.716 | 113.716 | 291.273 | -0.000 | -0.000 | 0.000 |
| -23.999 | 23.999 | -0.000 | 42.306 | -0.000 | 0.000 |
| -0.051 | -0.051 | 0.017 | -0.000 | 30.197 | -11.560 |
| -0.000 | -0.000 | -0.000 | -0.000 | -23.999 | 63.309 |
| 230.159 | 144.885 | 113.025 | -16.053 | 0.000 | -0.000 |
| 144.885 | 230.159 | 113.025 | 16.053 | 0.000 | -0.000 |
| 115.291 | 115.291 | 292.585 | -0.000 | 0.000 | -0.000 |
| -23.999 | 24.000 | 0.001 | 42.306 | -0.000 | -0.000 |
| 0.001 | 0.000 | 0.001 | 0.000 | 42.306 | -23.999 |
| -0.006 | -0.006 | 0.003 | 0.000 | -20.305 | 47.735 |
| 230.164 | 144.891 | 113.019 | -16.052 | -0.000 | 0.000 |
| 129.263 | 255.881 | 111.441 | 23.999 | 0.000 | 0.000 |
| 111.591 | 111.591 | 295.954 | -0.000 | -0.000 | 0.000 |
| -23.999 | 23.999 | -0.001 | 42.307 | 0.000 | 0.000 |
| -0.001 | -0.000 | -0.001 | -0.000 | 42.306 | -23.999 |
| 0.006 | 0.006 | -0.003 | -0.000 | -20.305 | 47.735 |
| 255.633 | 129.102 | 111.197 | -24.034 | 0.000 | 0.000 |
| 129.102 | 255.633 | 111.197 | 24.034 | -0.000 | 0.000 |
| -1316.033 | -1316.033 | 980.880 | 0.000 | -0.000 | 0.000 |
| -1439.331 | -1405.841 | 722.709 | 34.225 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 42.172 | -24.034 |
| -1430.031 | -1430.031 | 687.607 | -0.000 | -22.328 | 56.100 |
| 1652.525 | 1565.422 | -609.257 | -16.766 | 0.000 | -0.000 |
| 1560.370 | 1672.571 | -573.846 | 22.328 | 0.000 | -0.000 |
| 111.308 | 111.308 | 295.666 | -0.000 | 0.000 | -0.000 |
| -24.034 | 24.034 | 0.000 | 42.172 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 42.172 | -24.034 |
| 1430.031 | 1430.031 | -687.607 | -0.000 | -22.328 | 56.100 |
| 99.410 | -12.778 | 182.580 | -22.327 | -0.000 | 0.000 |
| -12.778 | 99.410 | 182.580 | 22.327 | -0.000 | 0.000 |
| 111.308 | 111.308 | 295.666 | 0.000 | 0.000 | 0.000 |
| -165.398 | -120.744 | 68.793 | 39.970 | -0.000 | 0.000 |
| -143.071 | -143.071 | 68.793 | 0.000 | 39.970 | -22.327 |
| 0.000 | 0.000 | 0.000 | 0.000 | -24.034 | 63.266 |
| 385.725 | 273.543 | 44.908 | -22.326 | -0.000 | -0.000 |
| 129.102 | 255.633 | 111.197 | 24.034 | -0.000 | -0.000 |
| 256.982 | 256.982 | 224.520 | -0.000 | 0.000 | -0.000 |
| 126.951 | 157.162 | -73.309 | 32.538 | -0.000 | -0.000 |
| 143.071 | 143.071 | -68.793 | -0.000 | 39.970 | -22.327 |
| -0.000 | -0.000 | -0.000 | -0.000 | -24.034 | 63.266 |
| 215.128 | 129.766 | 120.391 | -16.358 | -0.000 | 0.000 |
| 129.769 | 215.133 | 120.389 | 16.358 | -0.000 | 0.000 |
| 111.309 | 111.309 | 295.669 | -0.000 | -0.000 | 0.000 |
| -37.257 | 7.229 | 7.220 | 39.861 | -0.000 | 0.000 |
| -15.013 | -15.014 | 7.220 | 0.000 | 39.861 | -22.243 |
| -13.783 | -13.783 | 7.046 | 0.000 | -16.333 | 42.676 |
| 255.632 | 129.102 | 111.196 | -24.034 | 0.000 | -0.000 |
| 129.102 | 255.632 | 111.196 | 24.034 | 0.000 | -0.000 |
| 111.307 | 111.307 | 295.663 | -0.000 | 0.000 | -0.000 |
| 1.265 | 27.053 | -7.463 | 30.263 | 0.000 | -0.000 |
| 15.013 | 15.014 | -7.220 | -0.000 | 39.861 | -22.243 |
| 13.783 | 13.783 | -7.046 | -0.000 | -16.333 | 42.676 |
| -2920.170 | -3471.729 | -112.071 | -0.000 | 0.000 | 0.001 |
| -4554.118 | -5452.802 | 81.156 | -0.000 | -0.000 | 0.003 |
| 171.958 | 206.031 | 504.839 | -0.000 | 0.000 | -0.000 |
| -5008.770 | -6090.230 | -122.594 | 93.939 | -0.000 | 0.012 |
| -5017.328 | -5996.682 | -103.824 | -0.000 | 60.817 | -0.006 |
| -4301.761 | -4483.510 | -173.408 | -0.000 | -0.000 | 40.716 |
| 3496.968 | 3777.992 | 462.313 | 0.000 | 0.000 | -0.001 |
| 4448.817 | 4765.279 | 383.046 | -0.000 | 0.000 | 0.000 |
| 171.958 | 206.031 | 504.839 | -0.000 | 0.000 | 0.000 |
| 5008.770 | 6090.230 | 122.594 | 93.939 | 0.000 | -0.012 |
| -0.000 | -0.000 | -0.000 | -0.000 | 61.350 | 0.000 |
| 4301.761 | 4483.510 | 173.408 | 0.000 | 0.000 | 40.716 |
| -183.724 | -435.212 | 162.231 | -0.000 | 0.000 | 0.000 |
| -284.429 | -168.244 | 192.386 | -0.000 | -0.000 | -0.000 |
| -145.518 | -155.331 | 475.370 | 0.000 | -0.000 | 0.000 |
| -430.177 | -448.349 | -17.339 | 93.899 | -0.000 | -0.000 |
| -320.854 | -362.487 | -28.720 | 0.000 | 60.909 | 0.000 |
| -429.790 | -448.072 | -17.395 | -0.000 | -0.000 | 40.707 |
| 323.706 | 176.707 | 173.686 | 0.000 | 0.000 | -0.000 |
| 177.978 | 335.616 | 209.030 | -0.000 | 0.000 | -0.000 |
| 171.957 | 206.031 | 504.838 | 0.000 | 0.000 | 0.000 |
| 430.177 | 448.349 | 17.339 | 93.899 | 0.000 | 0.000 |
| 320.854 | 362.487 | 28.720 | 0.000 | 60.909 | -0.000 |
| 429.790 | 448.072 | 17.395 | 0.000 | 0.000 | 40.707 |
| 243.834 | 99.315 | 173.862 | 0.000 | -0.000 | -0.000 |
| 177.981 | 335.623 | 209.034 | -0.000 | 0.000 | -0.000 |
| 143.160 | 170.900 | 501.250 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 96.636 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 61.350 | -0.000 |
| -30.884 | -34.891 | -2.764 | -0.000 | 0.000 | 41.252 |
| 326.700 | 192.237 | 177.372 | -0.000 | 0.000 | -0.000 |
| 195.127 | 338.911 | 211.619 | -0.000 | 0.000 | -0.000 |
| 207.499 | 243.373 | 506.920 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 96.636 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 61.350 | 0.000 |
| 30.884 | 34.891 | 2.764 | 0.000 | 0.000 | 41.252 |
| 510.441 | 303.255 | 285.812 | 0.000 | 0.000 | 0.000 |
| 189.421 | 435.569 | 242.584 | 0.000 | 0.000 | -0.000 |
| 599.937 | 574.066 | 623.215 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 113.289 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 56.127 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 61.490 |
| 335.370 | 189.421 | 185.643 | -0.000 | -0.000 | 0.000 |
| 189.422 | 435.569 | 242.584 | -0.000 | -0.000 | -0.000 |
| -32.698 | 105.075 | 443.763 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 113.289 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 56.127 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 61.490 |
| 335.370 | 189.421 | 185.642 | 0.000 | 0.000 | -0.000 |
| 189.421 | 435.569 | 242.584 | 0.000 | 0.000 | -0.000 |
| 212.651 | 259.935 | 549.995 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 113.289 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 56.127 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 61.490 |
| 335.371 | 189.422 | 185.643 | -0.000 | -0.000 | -0.000 |
| 135.787 | 373.387 | 235.267 | -0.000 | -0.000 | -0.000 |
| 185.644 | 242.585 | 541.051 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 113.289 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 56.127 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 61.490 |
| 335.368 | 189.417 | 185.638 | 0.000 | 0.000 | 0.000 |
| 189.417 | 435.567 | 242.580 | 0.000 | 0.000 | -0.000 |
| 185.639 | 242.580 | 541.047 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.001 | 0.001 | 113.289 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 56.127 | 0.000 |
| 3.855 | 3.080 | 0.746 | 0.000 | 0.000 | 30.193 |
| 279.127 | 138.389 | 189.608 | -0.000 | -0.000 | 0.000 |
| 141.223 | 361.582 | 246.180 | -0.000 | -0.000 | -0.000 |
| 185.648 | 242.589 | 541.054 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.001 | -0.001 | 113.289 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 56.127 | -0.000 |
| -5.282 | -4.784 | -0.603 | -0.000 | -0.000 | 30.343 |
| 323.670 | 173.313 | 170.928 | 0.000 | -0.000 | 0.000 |
| -588.623 | -642.453 | 113.047 | -0.000 | -0.000 | 0.002 |
| -570.607 | -717.624 | 407.609 | -0.000 | 0.000 | 0.002 |
| -980.351 | -1315.987 | -56.983 | 91.924 | 0.000 | 0.007 |
| -878.977 | -1030.181 | -66.996 | 0.000 | 61.863 | -0.001 |
| -742.375 | -917.694 | -88.979 | -0.000 | 0.000 | 37.999 |
| 323.670 | 173.313 | 170.928 | -0.000 | 0.000 | -0.000 |
| 174.890 | 317.382 | 202.965 | 0.000 | 0.000 | -0.000 |
| 1050.522 | 1231.518 | 563.511 | -0.000 | 0.000 | 0.001 |
| 980.351 | 1315.987 | 56.983 | 91.924 | -0.000 | -0.007 |
| 878.977 | 1030.181 | 66.996 | 0.000 | 61.863 | 0.001 |
| 742.374 | 917.694 | 88.979 | 0.000 | 0.000 | 37.995 |
| 213.107 | 59.824 | 163.605 | 0.000 | 0.000 | 0.000 |
| 174.890 | 317.382 | 202.965 | -0.000 | -0.000 | 0.000 |
| 169.108 | 199.569 | 497.446 | 0.000 | -0.000 | 0.000 |
| -98.310 | -138.410 | -6.108 | 91.248 | 0.000 | -0.001 |
| 0.000 | 0.000 | 0.000 | -0.000 | 62.334 | 0.000 |
| -83.495 | -99.515 | -7.491 | -0.000 | -0.000 | 42.285 |
| 376.999 | 249.189 | 179.108 | 0.000 | 0.000 | 0.000 |
| 174.890 | 317.382 | 202.965 | 0.000 | -0.000 | -0.000 |
| 246.153 | 291.983 | 505.488 | -0.000 | -0.000 | -0.000 |
| 98.310 | 138.410 | 6.108 | 91.248 | -0.000 | 0.001 |
| -0.000 | -0.000 | -0.000 | -0.000 | 62.334 | -0.000 |
| 83.494 | 99.514 | 7.491 | 0.000 | -0.000 | 42.285 |
| 323.671 | 173.312 | 170.927 | 0.000 | -0.000 | 0.000 |
| 114.854 | 216.647 | 204.418 | 0.000 | -0.000 | -0.000 |
| 164.276 | 191.176 | 495.845 | -0.000 | -0.000 | 0.000 |
| -6.840 | -8.454 | -0.819 | 91.241 | -0.000 | 0.000 |
| -7.351 | -9.088 | -0.881 | -0.000 | 61.840 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 71.468 |
| 294.172 | 151.455 | 172.655 | -0.000 | -0.000 | 0.000 |
| 135.049 | 232.039 | 205.460 | 0.000 | -0.000 | 0.000 |
| 178.763 | 210.389 | 497.541 | -0.000 | 0.000 | 0.000 |
| 6.840 | 8.454 | 0.819 | 91.241 | 0.000 | -0.000 |
| 7.351 | 9.088 | 0.881 | -0.000 | 61.840 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 71.468 |
| 90.712 | -13.201 | 12.475 | 0.000 | 0.000 | -0.000 |
| 89.095 | 160.045 | 91.951 | 0.000 | 0.000 | -0.000 |
| -26.724 | 43.481 | 86.840 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 60.592 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 43.489 | -0.000 |
| -104.183 | -48.285 | -52.763 | 0.000 | 0.000 | 47.872 |
| 233.934 | 89.549 | 75.792 | -0.000 | -0.000 | 0.000 |
| 220.703 | 245.394 | 163.694 | -0.000 | 0.000 | 0.000 |
| 181.118 | 139.775 | 191.699 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 60.592 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 43.489 | 0.000 |
| 104.183 | 48.285 | 52.763 | -0.000 | -0.000 | 47.872 |
| 218.075 | 84.924 | 70.545 | 0.000 | 0.000 | -0.000 |
| 89.095 | 160.045 | 91.951 | 0.000 | 0.000 | -0.000 |
| 76.683 | 93.296 | 163.026 | 0.000 | 0.000 | -0.000 |
| -12.638 | -9.435 | -5.441 | 54.155 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 43.489 | -0.000 |
| -10.248 | -4.750 | -5.190 | 0.000 | 0.000 | 47.920 |
| 233.934 | 89.549 | 75.792 | -0.000 | -0.000 | 0.000 |
| 98.237 | 153.282 | 94.797 | -0.000 | -0.000 | 0.000 |
| 76.683 | 93.296 | 163.025 | -0.000 | -0.000 | 0.000 |
| 12.638 | 9.435 | 5.441 | 54.155 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 43.489 | -0.000 |
| 10.248 | 4.750 | 5.190 | -0.000 | -0.000 | 47.920 |
| 233.937 | 89.550 | 75.793 | 0.000 | 0.000 | -0.000 |
| 86.986 | 142.514 | 96.800 | 0.000 | 0.000 | -0.000 |
| 76.684 | 93.297 | 163.028 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 60.592 | 0.000 | -0.000 |
| -1.372 | -1.371 | -0.498 | 0.000 | 31.676 | -0.000 |
| -1.286 | -1.126 | -0.509 | 0.000 | 0.000 | 47.531 |
| 229.238 | 89.841 | 75.937 | -0.000 | -0.000 | 0.000 |
| 89.876 | 149.730 | 90.615 | 0.000 | 0.000 | 0.000 |
| 77.174 | 92.157 | 146.059 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 60.592 | -0.000 | 0.000 |
| 1.372 | 1.371 | 0.498 | -0.000 | 31.676 | 0.000 |
| 1.286 | 1.126 | 0.509 | -0.000 | 0.000 | 47.531 |
| 406.190 | 228.782 | 397.712 | 0.000 | -0.000 | -0.000 |
| 165.303 | 120.834 | 78.279 | 0.000 | -0.000 | 0.000 |
| 267.830 | 236.435 | 445.226 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 60.450 | 0.000 | -0.000 |
| 197.427 | 132.352 | 328.597 | 0.000 | 33.303 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 50.696 |
| 232.832 | 88.911 | 75.187 | -0.000 | 0.000 | -0.000 |
| 88.457 | 159.122 | 91.390 | -0.000 | 0.000 | 0.000 |
| -22.605 | 118.465 | 136.119 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 60.450 | 0.000 | 0.000 |
| -197.427 | -132.352 | -328.597 | -0.000 | 33.303 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 50.696 |
| 243.283 | 101.782 | 101.517 | -0.000 | -0.000 | -0.000 |
| 93.273 | 147.732 | 89.032 | 0.000 | -0.000 | -0.000 |
| 76.112 | 92.770 | 160.992 | 0.000 | -0.000 | -0.000 |
| 18.015 | 14.014 | 32.876 | 55.850 | -0.000 | 0.000 |
| 19.976 | 13.470 | 33.395 | -0.000 | 33.178 | -0.000 |
| 18.475 | 12.912 | 31.732 | 0.000 | -0.000 | 44.765 |
| 232.832 | 88.911 | 75.187 | -0.000 | 0.000 | 0.000 |
| 88.456 | 159.122 | 91.390 | 0.000 | 0.000 | 0.000 |
| 55.642 | 91.911 | 88.855 | 0.000 | 0.000 | 0.000 |
| -18.015 | -14.014 | -32.876 | 55.850 | 0.000 | -0.000 |
| -19.976 | -13.470 | -33.395 | 0.000 | 33.178 | -0.000 |
| -18.475 | -12.912 | -31.732 | -0.000 | 0.000 | 44.765 |
| 228.056 | 88.642 | 74.792 | 0.000 | -0.000 | -0.000 |
| 88.826 | 144.451 | 97.146 | -0.000 | -0.000 | -0.000 |
| 74.190 | 99.515 | 133.623 | 0.000 | -0.000 | -0.000 |
| 0.611 | -0.181 | -0.083 | 55.938 | -0.000 | -0.000 |
| 2.183 | 1.919 | 4.482 | 0.000 | 31.832 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 50.696 |
| 226.045 | 88.934 | 74.877 | -0.000 | 0.000 | 0.000 |
| 88.456 | 159.121 | 91.389 | -0.000 | 0.000 | 0.000 |
| 75.269 | 91.200 | 141.411 | 0.000 | 0.000 | 0.000 |
| -0.611 | 0.181 | 0.083 | 55.938 | 0.000 | 0.000 |
| -2.183 | -1.919 | -4.482 | 0.000 | 31.832 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 50.696 |
| 492.173 | 260.201 | 244.573 | -6.162 | 0.000 | -0.000 |
| 260.201 | 492.173 | 244.573 | 6.162 | 0.000 | 0.000 |
| 244.577 | 244.577 | 454.168 | 0.000 | -0.000 | 0.000 |
| -14954.893 | -14904.133 | -12045.548 | 44.607 | 0.000 | 0.000 |
| -14929.513 | -14929.513 | -12045.548 | 0.000 | 44.607 | -25.380 |
| -29786.701 | -29786.701 | -31288.486 | -0.000 | -21.666 | 87.537 |
| 492.173 | 260.201 | 244.573 | -6.162 | -0.000 | -0.000 |
| 260.201 | 492.173 | 244.573 | 6.162 | 0.000 | 0.000 |
| 244.577 | 244.577 | 454.168 | 0.000 | -0.000 | 0.000 |
| 8831.132 | 8855.486 | 6405.553 | 89.640 | 0.000 | -0.000 |
| 14929.513 | 14929.513 | 12045.548 | -0.000 | 44.607 | -25.380 |
| 29786.701 | 29786.701 | 31288.486 | 0.000 | -21.666 | 87.537 |
| -1311.938 | -1427.783 | -1002.242 | -25.417 | -0.000 | 0.000 |
| -1427.783 | -1311.938 | -1002.242 | 25.417 | 0.000 | 0.000 |
| -1355.260 | -1355.260 | -953.296 | 0.000 | 0.000 | -0.000 |
| -6.162 | 6.162 | 0.000 | 104.869 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 104.869 | -6.162 |
| -1491.001 | -1491.001 | -1203.014 | -0.000 | -25.357 | 58.321 |
| 1669.452 | 1550.888 | 1402.049 | -25.122 | 0.000 | -0.000 |
| 1046.029 | 1232.328 | 822.781 | 12.510 | -0.000 | -0.000 |
| 244.577 | 244.577 | 454.168 | 0.000 | -0.000 | -0.000 |
| -6.162 | 6.162 | -0.000 | 104.869 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 104.869 | -6.162 |
| 1491.001 | 1491.001 | 1203.014 | 0.000 | -25.357 | 58.321 |
| 492.178 | 260.205 | 244.574 | -6.162 | 0.000 | 0.000 |
| 260.205 | 492.178 | 244.574 | 6.162 | -0.000 | 0.000 |
| 117.230 | 117.230 | 332.642 | -0.000 | -0.000 | 0.000 |
| -6.162 | 6.162 | 0.000 | 104.869 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 104.869 | -6.162 |
| 0.000 | 0.000 | 0.000 | 0.000 | -6.162 | 115.986 |
| 403.339 | 212.038 | 217.702 | -11.814 | 0.000 | -0.000 |
| 256.322 | 417.617 | 311.590 | 18.007 | 0.000 | -0.000 |
| 244.577 | 244.577 | 454.169 | -0.000 | 0.000 | -0.000 |
| 76.372 | 100.743 | 64.146 | 89.619 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 104.869 | -6.162 |
| -0.000 | -0.000 | -0.000 | -0.000 | -6.162 | 115.986 |
| 460.994 | 250.976 | 244.679 | -8.833 | -0.000 | -0.000 |
| -309.357 | -142.300 | 374.687 | 15.782 | 0.000 | 0.000 |
| 245.044 | 245.044 | 414.622 | 0.000 | -0.000 | -0.000 |
| -5548.643 | -5522.027 | -5129.505 | 86.436 | -0.000 | -0.000 |
| -1310.882 | -1310.882 | 1808.091 | 0.000 | 69.084 | -17.094 |
| -0.000 | 0.000 | -0.000 | 0.000 | -8.833 | 105.009 |
| 5860.471 | 5683.378 | 5300.600 | -13.349 | 0.000 | 0.000 |
| 250.977 | 460.994 | 244.679 | 8.833 | -0.000 | 0.000 |
| 5710.984 | 5710.984 | 5457.174 | -0.000 | -0.000 | 0.000 |
| 5522.027 | 5548.643 | 5129.505 | 86.436 | -0.000 | 0.000 |
| 1310.882 | 1310.882 | -1808.091 | -0.000 | 69.084 | -17.094 |
| 0.000 | 0.000 | 0.000 | -0.000 | -8.833 | 105.009 |
| 460.994 | 250.976 | 244.678 | -8.833 | -0.000 | -0.000 |
| -395.278 | -215.136 | -334.411 | 12.930 | -0.000 | -0.000 |
| -371.767 | -371.767 | -177.531 | 0.000 | -0.000 | 0.000 |
| -8.833 | 8.833 | -0.000 | 99.958 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 99.958 | -8.833 |
| -42.439 | -42.439 | 23.124 | -0.000 | -15.784 | 83.547 |
| 460.994 | 250.977 | 244.679 | -8.833 | 0.000 | 0.000 |
| 250.977 | 460.994 | 244.679 | 8.833 | 0.000 | 0.000 |
| 245.045 | 245.045 | 414.624 | -0.000 | 0.000 | 0.000 |
| -292.885 | -242.418 | -1032.720 | 55.874 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 99.958 | -8.833 |
| 42.439 | 42.439 | -23.124 | 0.000 | -15.784 | 83.547 |
| 311.902 | 122.135 | 138.694 | -11.610 | -0.000 | -0.000 |
| 250.973 | 460.992 | 244.674 | 8.833 | -0.000 | -0.000 |
| 245.038 | 245.038 | 414.616 | 0.000 | -0.000 | -0.000 |
| -8.833 | 8.833 | 0.000 | 99.958 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 99.958 | -8.833 |
| -54.576 | -54.576 | -50.575 | -0.000 | -13.245 | 88.925 |
| 460.996 | 250.980 | 244.684 | -8.833 | 0.000 | 0.000 |
| 250.980 | 460.996 | 244.684 | 8.833 | 0.000 | -0.000 |
| 161.687 | 161.687 | 308.201 | 0.000 | 0.000 | 0.000 |
| -8.833 | 8.833 | -0.000 | 99.958 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 99.958 | -8.833 |
| 54.576 | 54.576 | 50.575 | 0.000 | -13.245 | 88.925 |
| 180.387 | -1613.647 | 572.682 | 0.000 | -0.000 | 0.000 |
| -1925.539 | -1623.569 | 3475.445 | 0.000 | 0.000 | -0.000 |
| 31.649 | -1619.853 | 746.031 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 86.169 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 99.160 | 0.000 |
| -121.122 | -1773.045 | 420.436 | -0.000 | 0.000 | 84.606 |
| 342.227 | 170.846 | 167.391 | -0.000 | 0.000 | -0.000 |
| 170.287 | 377.947 | 160.625 | 0.000 | -0.000 | -0.000 |
| 1736.410 | 2241.327 | -1797.956 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 86.169 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 99.160 | 0.000 |
| 121.122 | 1773.045 | -420.436 | 0.000 | -0.000 | 84.606 |
| 289.194 | -18.063 | 194.241 | 0.000 | 0.000 | -0.000 |
| 170.287 | 377.947 | 160.626 | -0.000 | 0.000 | -0.000 |
| -44.157 | -64.867 | 319.485 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 86.169 | -0.000 | -0.000 |
| -12.114 | -177.326 | 42.049 | -0.000 | 89.415 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 95.581 |
| 342.227 | 170.846 | 167.391 | -0.000 | 0.000 | -0.000 |
| 170.287 | 377.947 | 160.625 | 0.000 | 0.000 | -0.000 |
| 304.570 | 332.586 | 216.977 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 86.169 | 0.000 | -0.000 |
| 12.114 | 177.326 | -42.049 | 0.000 | 89.415 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 95.581 |
| 217.927 | 120.747 | 136.504 | 0.000 | -0.000 | 0.000 |
| 170.223 | 377.897 | 160.590 | 0.000 | 0.000 | 0.000 |
| 150.727 | 133.870 | 328.519 | 0.000 | -0.000 | -0.000 |
| -17.340 | -19.655 | 4.996 | 53.643 | -0.000 | -0.000 |
| -1.226 | -17.946 | 4.256 | 0.000 | 89.298 | -0.000 |
| -0.003 | -0.003 | -0.002 | 0.000 | -0.000 | 95.580 |
| 247.073 | 157.050 | 126.384 | 0.000 | 0.000 | 0.000 |
| 160.102 | 382.129 | 146.339 | 0.000 | 0.000 | -0.000 |
| 168.083 | 161.875 | 357.764 | 0.000 | 0.000 | 0.000 |
| 17.340 | 19.655 | -4.996 | 53.643 | 0.000 | -0.000 |
| 1.226 | 17.946 | -4.256 | -0.000 | 89.298 | -0.000 |
| 0.003 | 0.003 | 0.002 | 0.000 | -0.000 | 95.580 |
| 553.204 | -335.042 | -745.705 | 0.000 | -0.000 | 0.000 |
| 4095.641 | 7451.527 | 1667.956 | 0.000 | -0.000 | 0.000 |
| 161.707 | 154.686 | 349.932 | 0.000 | -0.000 | 0.000 |
| 3617.522 | 3214.354 | 4519.508 | 50.735 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 95.245 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 91.383 |
| -1590.901 | -2446.743 | -2018.532 | -0.000 | -0.000 | -0.000 |
| -1684.734 | -2257.742 | -2021.794 | -0.000 | 0.000 | -0.000 |
| -1686.382 | -2448.911 | -1870.191 | -0.000 | 0.000 | -0.000 |
| -3617.522 | -3214.354 | -4519.508 | 50.735 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 95.245 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 91.383 |
| 299.733 | 99.545 | 51.145 | 0.000 | -0.000 | 0.000 |
| 486.522 | 633.790 | 573.716 | -0.000 | -0.000 | 0.000 |
| 307.039 | 384.813 | 488.240 | -0.000 | 0.000 | 0.000 |
| 361.785 | 321.456 | 451.997 | 50.734 | -0.000 | 0.000 |
| 348.099 | 651.081 | 142.201 | 0.000 | 72.714 | 0.000 |
| 326.464 | 519.908 | 212.259 | 0.000 | -0.000 | 61.715 |
| 39.937 | -127.993 | -88.579 | -0.000 | 0.000 | -0.000 |
| -237.267 | -9.485 | -330.390 | -0.000 | 0.000 | -0.000 |
| -55.364 | -130.298 | 59.521 | -0.000 | 0.000 | -0.000 |
| -361.785 | -321.456 | -451.997 | 50.734 | 0.000 | -0.000 |
| -181.219 | -257.615 | -214.406 | -0.000 | 72.984 | -0.000 |
| -326.464 | -519.908 | -212.259 | -0.000 | 0.000 | 61.715 |
| 258.366 | 159.123 | 175.475 | 0.000 | -0.000 | 0.000 |
| 147.374 | 346.318 | 145.467 | 0.000 | -0.000 | 0.000 |
| 144.282 | 152.767 | 295.942 | 0.000 | -0.000 | 0.000 |
| 36.501 | 32.349 | 45.654 | 50.626 | -0.000 | -0.000 |
| 36.178 | 32.076 | 45.183 | 0.000 | 72.146 | 0.000 |
| 32.650 | 51.932 | 21.322 | 0.000 | -0.000 | 61.682 |
| 204.692 | 104.859 | 105.104 | 0.000 | -0.000 | -0.000 |
| 87.213 | 278.139 | 75.672 | -0.000 | 0.000 | -0.000 |
| 111.099 | 79.992 | 268.225 | -0.000 | -0.000 | -0.000 |
| -36.501 | -32.349 | -45.654 | 50.626 | 0.000 | -0.000 |
| -18.092 | -25.739 | -21.433 | 0.000 | 73.041 | -0.000 |
| -32.650 | -51.932 | -21.322 | -0.000 | 0.000 | 61.682 |
| 1353.128 | 335.930 | 335.930 | 0.000 | 0.000 | 0.000 |
| 335.930 | 1353.128 | 335.930 | -0.000 | 0.000 | 0.000 |
| 335.930 | 335.930 | 1353.128 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -28.420 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -28.420 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -28.420 |
| 1353.129 | 335.931 | 335.931 | 0.000 | -0.000 | -0.000 |
| 335.931 | 1353.129 | 335.931 | 0.000 | 0.000 | -0.000 |
| 335.931 | 335.931 | 1353.129 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -28.420 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -28.420 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -28.420 |
| 1353.126 | 335.927 | 335.927 | -0.000 | -0.000 | 0.000 |
| 335.927 | 1353.126 | 335.927 | -0.000 | -0.000 | 0.000 |
| 335.927 | 335.927 | 1353.126 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -28.420 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -28.420 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -28.420 |
| 1353.131 | 335.934 | 335.934 | 0.000 | 0.000 | 0.000 |
| 335.934 | 1353.131 | 335.934 | 0.000 | -0.000 | -0.000 |
| 335.934 | 335.934 | 1353.131 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -28.420 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -28.420 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -28.420 |
| 1353.108 | 335.899 | 335.899 | 0.000 | 0.000 | -0.000 |
| 335.899 | 1353.108 | 335.899 | 0.000 | 0.000 | 0.000 |
| 335.899 | 335.899 | 1353.108 | 0.000 | 0.000 | 0.000 |
| 0.001 | 0.002 | 0.002 | -28.420 | 0.000 | 0.000 |
| 0.002 | 0.001 | 0.002 | 0.000 | -28.420 | 0.000 |
| 0.002 | 0.002 | 0.001 | 0.000 | -0.000 | -28.420 |
| 1353.149 | 335.962 | 335.962 | -0.000 | -0.000 | -0.000 |
| 335.962 | 1353.149 | 335.962 | -0.000 | 0.000 | -0.000 |
| 335.962 | 335.962 | 1353.149 | -0.000 | -0.000 | 0.000 |
| -0.001 | -0.002 | -0.002 | -28.420 | -0.000 | -0.000 |
| -0.002 | -0.001 | -0.002 | -0.000 | -28.420 | -0.000 |
| -0.002 | -0.002 | -0.001 | -0.000 | -0.000 | -28.420 |
| 33.546 | 0.856 | 0.856 | -0.000 | -0.000 | -0.000 |
| 0.856 | 33.546 | 0.856 | -0.000 | -0.000 | 0.000 |
| 0.856 | 0.856 | 33.546 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -2.036 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -2.036 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -2.036 |
| 33.546 | 0.856 | 0.856 | 0.000 | 0.000 | 0.000 |
| 0.856 | 33.546 | 0.856 | 0.000 | -0.000 | -0.000 |
| 0.856 | 0.856 | 33.546 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -2.036 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -2.036 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -2.036 |
| 33.546 | 0.856 | 0.856 | -0.000 | -0.000 | -0.000 |
| 0.856 | 33.546 | 0.856 | -0.000 | 0.000 | 0.000 |
| 0.856 | 0.856 | 33.546 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -2.036 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -2.036 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -2.036 |
| 33.546 | 0.856 | 0.856 | 0.000 | 0.000 | 0.000 |
| 0.856 | 33.546 | 0.856 | 0.000 | 0.000 | 0.000 |
| 0.856 | 0.856 | 33.546 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -2.036 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -2.036 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -2.036 |
| 33.546 | 0.856 | 0.856 | -0.000 | -0.000 | -0.000 |
| 0.856 | 33.546 | 0.856 | -0.000 | -0.000 | -0.000 |
| 0.856 | 0.856 | 33.546 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -2.036 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -2.036 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -2.036 |
| 33.546 | 0.856 | 0.856 | 0.000 | -0.000 | 0.000 |
| 0.856 | 33.546 | 0.856 | 0.000 | -0.000 | -0.000 |
| 0.856 | 0.856 | 33.546 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -2.036 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -2.036 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -2.036 |
| 400.976 | 346.843 | 346.843 | 0.000 | 0.000 | -0.000 |
| 346.843 | 400.976 | 346.843 | 0.000 | 0.000 | 0.000 |
| 346.843 | 346.843 | 400.976 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 222.356 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 222.356 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 222.356 |
| 400.976 | 346.843 | 346.843 | -0.000 | -0.000 | -0.000 |
| 346.843 | 400.976 | 346.843 | -0.000 | -0.000 | -0.000 |
| 346.843 | 346.843 | 400.976 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 222.356 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 222.356 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 222.356 |
| 400.976 | 346.843 | 346.843 | 0.000 | 0.000 | 0.000 |
| 346.843 | 400.976 | 346.843 | 0.000 | 0.000 | 0.000 |
| 346.843 | 346.843 | 400.976 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 222.356 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 222.356 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 222.356 |
| 400.975 | 346.842 | 346.842 | -0.000 | -0.000 | 0.000 |
| 346.842 | 400.975 | 346.842 | -0.000 | -0.000 | 0.000 |
| 346.842 | 346.842 | 400.975 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 222.356 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 222.356 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 222.356 |
| 400.981 | 346.848 | 346.848 | 0.000 | -0.000 | 0.000 |
| 346.848 | 400.981 | 346.848 | 0.000 | 0.000 | 0.000 |
| 346.848 | 346.848 | 400.981 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.001 | -0.000 | 222.356 | 0.000 | 0.000 |
| -0.001 | 0.000 | -0.000 | 0.000 | 222.356 | -0.000 |
| -0.001 | -0.000 | 0.000 | 0.000 | -0.000 | 222.356 |
| 400.970 | 346.837 | 346.837 | -0.000 | -0.000 | -0.000 |
| 346.837 | 400.970 | 346.837 | -0.000 | 0.000 | -0.000 |
| 346.837 | 346.837 | 400.970 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.001 | 0.000 | 222.356 | -0.000 | -0.000 |
| 0.001 | -0.000 | 0.000 | -0.000 | 222.356 | -0.000 |
| 0.001 | 0.000 | -0.000 | -0.000 | -0.000 | 222.356 |
| 50.300 | 22.598 | 22.598 | 0.000 | 0.000 | 0.000 |
| 22.598 | 50.300 | 22.598 | 0.000 | 0.000 | 0.000 |
| 22.598 | 22.598 | 50.300 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 17.100 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 17.100 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 17.100 |
| 50.300 | 22.598 | 22.598 | -0.000 | -0.000 | -0.000 |
| 22.598 | 50.300 | 22.598 | -0.000 | -0.000 | -0.000 |
| 22.598 | 22.598 | 50.300 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 17.100 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 17.100 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 17.100 |
| 50.300 | 22.598 | 22.598 | 0.000 | 0.000 | 0.000 |
| 22.598 | 50.300 | 22.598 | 0.000 | 0.000 | 0.000 |
| 22.598 | 22.598 | 50.300 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 17.100 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 17.100 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 17.100 |
| 50.300 | 22.598 | 22.598 | -0.000 | -0.000 | -0.000 |
| 22.598 | 50.300 | 22.598 | -0.000 | -0.000 | -0.000 |
| 22.598 | 22.598 | 50.300 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 17.100 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 17.100 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 17.100 |
| 50.300 | 22.598 | 22.598 | 0.000 | 0.000 | 0.000 |
| 22.598 | 50.300 | 22.598 | 0.000 | 0.000 | 0.000 |
| 22.598 | 22.598 | 50.300 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 17.100 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 17.100 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 17.100 |
| 50.300 | 22.598 | 22.598 | -0.000 | -0.000 | -0.000 |
| 22.598 | 50.300 | 22.598 | -0.000 | -0.000 | -0.000 |
| 22.598 | 22.598 | 50.300 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 17.100 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 17.100 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 17.100 |
| 44.366 | 60.214 | 60.214 | 0.000 | 0.000 | 0.000 |
| 60.214 | 44.366 | 60.214 | 0.000 | 0.000 | 0.000 |
| 60.214 | 60.214 | 44.366 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 45.924 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 45.924 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 45.924 |
| 44.366 | 60.214 | 60.214 | -0.000 | -0.000 | -0.000 |
| 60.214 | 44.366 | 60.214 | -0.000 | -0.000 | -0.000 |
| 60.214 | 60.214 | 44.366 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 45.924 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 45.924 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 45.924 |
| 44.366 | 60.214 | 60.214 | 0.000 | 0.000 | 0.000 |
| 60.214 | 44.366 | 60.214 | 0.000 | 0.000 | 0.000 |
| 60.214 | 60.214 | 44.366 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 45.924 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 45.924 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 45.924 |
| 44.366 | 60.214 | 60.214 | -0.000 | -0.000 | -0.000 |
| 60.214 | 44.366 | 60.214 | -0.000 | -0.000 | -0.000 |
| 60.214 | 60.214 | 44.366 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 45.924 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 45.924 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 45.924 |
| 44.365 | 60.214 | 60.214 | 0.000 | 0.000 | 0.000 |
| 60.214 | 44.365 | 60.214 | 0.000 | 0.000 | 0.000 |
| 60.214 | 60.214 | 44.365 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 45.924 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 45.924 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 45.924 |
| 44.366 | 60.214 | 60.214 | -0.000 | -0.000 | -0.000 |
| 60.214 | 44.366 | 60.214 | -0.000 | -0.000 | -0.000 |
| 60.214 | 60.214 | 44.366 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 45.924 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 45.924 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 45.924 |
| 6.770 | 6.770 | 6.770 | 0.000 | 0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | 0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | -0.000 | 0.000 | -0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | -0.000 | -0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | -0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | 0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | -0.000 | 0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | -0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | -0.000 | -0.000 | -0.000 |
| 6.770 | 6.770 | 6.770 | -0.000 | 0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | -0.000 | 0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | -0.000 | 0.000 |
| 6.770 | 6.770 | 6.770 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| -81.925 | 309.600 | 309.600 | 0.000 | 0.000 | 0.000 |
| 309.600 | -81.925 | 309.600 | 0.000 | 0.000 | 0.000 |
| 309.600 | 309.600 | -81.925 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -199.812 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -199.812 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -199.812 |
| -81.926 | 309.600 | 309.600 | -0.000 | -0.000 | -0.000 |
| 309.600 | -81.926 | 309.600 | -0.000 | -0.000 | -0.000 |
| 309.600 | 309.600 | -81.926 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -199.812 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -199.812 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -199.812 |
| -81.925 | 309.600 | 309.600 | 0.000 | 0.000 | 0.000 |
| 309.600 | -81.925 | 309.600 | 0.000 | 0.000 | 0.000 |
| 309.600 | 309.600 | -81.925 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -199.812 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -199.812 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -199.812 |
| -81.926 | 309.600 | 309.600 | -0.000 | -0.000 | -0.000 |
| 309.600 | -81.926 | 309.600 | -0.000 | -0.000 | -0.000 |
| 309.600 | 309.600 | -81.926 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -199.812 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -199.812 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -199.812 |
| -81.919 | 309.603 | 309.603 | 0.000 | 0.000 | 0.000 |
| 309.603 | -81.919 | 309.603 | 0.000 | 0.000 | 0.000 |
| 309.603 | 309.603 | -81.919 | 0.000 | 0.000 | 0.000 |
| 0.001 | 0.000 | 0.000 | -199.812 | 0.000 | 0.000 |
| 0.000 | 0.001 | 0.000 | 0.000 | -199.812 | 0.000 |
| 0.000 | 0.000 | 0.001 | 0.000 | 0.000 | -199.812 |
| -81.932 | 309.597 | 309.597 | -0.000 | -0.000 | -0.000 |
| 309.597 | -81.932 | 309.597 | -0.000 | -0.000 | -0.000 |
| 309.597 | 309.597 | -81.932 | -0.000 | -0.000 | -0.000 |
| -0.001 | -0.000 | -0.000 | -199.812 | -0.000 | -0.000 |
| -0.000 | -0.001 | -0.000 | -0.000 | -199.812 | -0.000 |
| -0.000 | -0.000 | -0.001 | -0.000 | -0.000 | -199.812 |
| 56.182 | 56.896 | 56.896 | -0.000 | -0.000 | 0.000 |
| 56.896 | 56.182 | 56.896 | -0.000 | -0.000 | 0.000 |
| 56.896 | 56.896 | 56.182 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 3.239 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 3.239 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 3.239 |
| 56.182 | 56.896 | 56.896 | 0.000 | 0.000 | -0.000 |
| 56.896 | 56.182 | 56.896 | 0.000 | -0.000 | -0.000 |
| 56.896 | 56.896 | 56.182 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 3.239 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 3.239 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 3.239 |
| 56.182 | 56.896 | 56.896 | -0.000 | -0.000 | 0.000 |
| 56.896 | 56.182 | 56.896 | -0.000 | -0.000 | 0.000 |
| 56.896 | 56.896 | 56.182 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 3.239 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 3.239 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 3.239 |
| 56.182 | 56.896 | 56.896 | 0.000 | 0.000 | -0.000 |
| 56.896 | 56.182 | 56.896 | 0.000 | 0.000 | 0.000 |
| 56.896 | 56.896 | 56.182 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 3.239 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 3.239 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 3.239 |
| 56.182 | 56.896 | 56.896 | -0.000 | -0.000 | 0.000 |
| 56.896 | 56.182 | 56.896 | -0.000 | -0.000 | 0.000 |
| 56.896 | 56.896 | 56.182 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -13.627 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -13.627 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -13.627 |
| 56.182 | 56.896 | 56.896 | 0.000 | -0.000 | -0.000 |
| 56.896 | 56.182 | 56.896 | -0.000 | 0.000 | -0.000 |
| 56.896 | 56.896 | 56.182 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -13.627 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -13.627 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -13.627 |
| 55.767 | 24.242 | 24.242 | -0.000 | -0.000 | 0.000 |
| 24.242 | 55.767 | 24.242 | -0.000 | -0.000 | 0.000 |
| 24.242 | 24.242 | 55.767 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 8.466 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 8.466 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 8.466 |
| 55.767 | 24.242 | 24.242 | 0.000 | 0.000 | -0.000 |
| 24.242 | 55.767 | 24.242 | 0.000 | 0.000 | 0.000 |
| 24.242 | 24.242 | 55.767 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 8.466 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 8.466 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 8.466 |
| 55.767 | 24.242 | 24.242 | -0.000 | -0.000 | 0.000 |
| 24.242 | 55.767 | 24.242 | -0.000 | -0.000 | 0.000 |
| 24.242 | 24.242 | 55.767 | -0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 8.466 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 8.466 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 8.466 |
| 55.767 | 24.242 | 24.242 | 0.000 | 0.000 | -0.000 |
| 24.242 | 55.767 | 24.242 | 0.000 | 0.000 | -0.000 |
| 24.242 | 24.242 | 55.767 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 8.466 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 8.466 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 8.466 |
| 55.767 | 24.243 | 24.243 | -0.000 | -0.000 | -0.000 |
| 24.243 | 55.767 | 24.243 | -0.000 | -0.000 | 0.000 |
| 24.243 | 24.243 | 55.767 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 11.115 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 11.115 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 11.115 |
| 55.766 | 24.241 | 24.241 | 0.000 | -0.000 | -0.000 |
| 24.241 | 55.766 | 24.241 | -0.000 | 0.000 | -0.000 |
| 24.241 | 24.241 | 55.766 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 11.115 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 11.115 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 11.115 |
| 395.311 | 477.428 | 477.428 | -0.000 | -0.000 | -0.000 |
| 477.428 | 395.311 | 477.428 | -0.000 | -0.000 | -0.000 |
| 477.428 | 477.428 | 395.311 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 316.739 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 316.739 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 316.739 |
| 395.311 | 477.428 | 477.428 | 0.000 | 0.000 | -0.000 |
| 477.428 | 395.311 | 477.428 | 0.000 | 0.000 | -0.000 |
| 477.428 | 477.428 | 395.311 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 316.739 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 316.739 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 316.739 |
| 395.311 | 477.428 | 477.428 | -0.000 | -0.000 | -0.000 |
| 477.428 | 395.311 | 477.428 | -0.000 | -0.000 | -0.000 |
| 477.428 | 477.428 | 395.311 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 316.739 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 316.739 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 316.739 |
| 395.311 | 477.428 | 477.428 | 0.000 | 0.000 | 0.000 |
| 477.428 | 395.311 | 477.428 | 0.000 | 0.000 | 0.000 |
| 477.428 | 477.428 | 395.311 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 316.739 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 316.739 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 316.739 |
| 395.312 | 477.430 | 477.430 | -0.000 | -0.000 | -0.000 |
| 477.430 | 395.312 | 477.430 | -0.000 | -0.000 | -0.000 |
| 477.430 | 477.430 | 395.312 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 205.272 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 205.272 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 205.272 |
| 395.310 | 477.427 | 477.427 | 0.000 | 0.000 | 0.000 |
| 477.427 | 395.310 | 477.427 | 0.000 | 0.000 | 0.000 |
| 477.427 | 477.427 | 395.310 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 205.272 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 205.272 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 205.272 |
| -628.473 | -547.499 | -547.499 | -0.000 | -0.000 | -0.000 |
| -547.499 | -628.473 | -547.499 | 0.000 | -0.000 | -0.000 |
| -547.499 | -547.499 | -628.473 | 0.000 | -0.000 | -0.000 |
| -0.012 | -0.012 | -0.012 | -473.085 | 0.000 | 0.000 |
| -0.012 | -0.012 | -0.012 | 0.000 | -473.085 | -0.000 |
| -0.012 | -0.012 | -0.012 | -0.000 | -0.000 | -473.085 |
| -628.450 | -547.476 | -547.476 | 0.000 | 0.000 | 0.000 |
| -547.476 | -628.450 | -547.476 | 0.000 | 0.000 | 0.000 |
| -547.476 | -547.476 | -628.450 | 0.000 | 0.000 | 0.000 |
| 0.012 | 0.012 | 0.012 | -473.085 | 0.000 | -0.000 |
| 0.012 | 0.012 | 0.012 | -0.000 | -473.085 | 0.000 |
| 0.012 | 0.012 | 0.012 | 0.000 | 0.000 | -473.085 |
| -219496.904 | -219497.052 | -219439.558 | -466.278 | -468.232 | -414.637 |
| -219673.728 | -219672.669 | -219615.746 | -432.940 | -499.880 | -415.171 |
| -219491.926 | -219491.464 | -219433.521 | -439.996 | -471.095 | -407.142 |
| -3899.268 | -4126.434 | -4126.625 | -956876.620 | -0.001 | -0.000 |
| -4126.434 | -3899.268 | -4126.625 | -0.001 | -956876.620 | -0.000 |
| -4126.434 | -4126.625 | -3899.268 | -0.000 | -0.001 | -956876.619 |
| -628.449 | -547.476 | -547.476 | -0.000 | -0.000 | 0.000 |
| -547.476 | -628.449 | -547.476 | -0.000 | 0.000 | 0.000 |
| -547.476 | -547.476 | -628.449 | -0.000 | -0.000 | 0.000 |
| 3899.268 | 4126.434 | 4126.625 | -956876.618 | 0.001 | 0.000 |
| 4126.434 | 3899.268 | 4126.625 | 0.001 | -956876.618 | 0.000 |
| 4126.434 | 4126.625 | 3899.268 | -0.000 | 0.001 | -956876.619 |
| -628.477 | -547.500 | -547.500 | 0.000 | -0.000 | -0.000 |
| -547.500 | -628.477 | -547.500 | 0.000 | -0.000 | 0.000 |
| -547.500 | -547.500 | -628.477 | 0.000 | -0.000 | 0.000 |
| -10698.628 | -9796.183 | -9797.139 | -478014.834 | -0.000 | -0.000 |
| -9796.183 | -10698.628 | -9797.139 | -0.000 | -478014.836 | -0.000 |
| -9796.183 | -9797.139 | -10698.628 | -0.000 | -0.000 | -478014.838 |
| -628.445 | -547.475 | -547.475 | -0.000 | 0.000 | -0.000 |
| -547.475 | -628.445 | -547.475 | -0.000 | 0.000 | 0.000 |
| -547.475 | -547.475 | -628.445 | -0.000 | 0.000 | -0.000 |
| 10698.627 | 9796.183 | 9797.139 | -478014.833 | 0.000 | 0.000 |
| 9796.183 | 10698.627 | 9797.139 | 0.000 | -478014.833 | 0.000 |
| 9796.183 | 9797.139 | 10698.628 | 0.000 | 0.000 | -478014.840 |
| -347.017 | -339.288 | -339.288 | 0.000 | 0.000 | 0.000 |
| -339.288 | -347.017 | -339.288 | 0.000 | 0.000 | 0.000 |
| -339.288 | -339.288 | -347.017 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -340.190 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -340.190 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -340.190 |
| -347.017 | -339.288 | -339.288 | -0.000 | -0.000 | -0.000 |
| -339.288 | -347.017 | -339.288 | -0.000 | -0.000 | -0.000 |
| -339.288 | -339.288 | -347.017 | -0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -340.190 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -340.190 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | -340.190 |
| -347.017 | -339.289 | -339.289 | -0.000 | -0.000 | 0.000 |
| -339.289 | -347.017 | -339.289 | -0.000 | -0.000 | 0.000 |
| -339.289 | -339.289 | -347.017 | -0.000 | -0.000 | 0.000 |
| -1191.808 | -7113.990 | -7114.721 | -365683.867 | -0.000 | -0.000 |
| -7113.990 | -1191.808 | -7114.721 | -0.000 | -365683.868 | -0.000 |
| -7113.990 | -7114.721 | -1191.808 | -0.000 | -0.000 | -365683.866 |
| -347.016 | -339.287 | -339.287 | -0.000 | -0.000 | -0.000 |
| -339.287 | -347.016 | -339.287 | -0.000 | 0.000 | -0.000 |
| -339.287 | -339.287 | -347.016 | -0.000 | -0.000 | -0.000 |
| 1191.808 | 7113.990 | 7114.722 | -365683.869 | 0.000 | 0.000 |
| 7113.990 | 1191.808 | 7114.721 | 0.000 | -365683.868 | 0.000 |
| 7113.990 | 7114.722 | 1191.808 | 0.000 | 0.000 | -365683.869 |
| 45.845 | 31.988 | 31.988 | -0.000 | -0.000 | -0.000 |
| 31.988 | 45.845 | 31.988 | -0.000 | -0.000 | 0.000 |
| 31.988 | 31.988 | 45.845 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 25.162 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 25.162 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 25.162 |
| 45.845 | 31.988 | 31.988 | 0.000 | 0.000 | 0.000 |
| 31.988 | 45.845 | 31.988 | 0.000 | 0.000 | 0.000 |
| 31.988 | 31.988 | 45.845 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 25.162 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 25.162 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 25.162 |
| 45.845 | 31.988 | 31.988 | -0.000 | -0.000 | -0.000 |
| 31.988 | 45.845 | 31.988 | -0.000 | -0.000 | -0.000 |
| 31.988 | 31.988 | 45.845 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 24.636 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 24.636 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 24.636 |
| 45.845 | 31.988 | 31.988 | 0.000 | 0.000 | 0.000 |
| 31.988 | 45.845 | 31.988 | 0.000 | 0.000 | -0.000 |
| 31.988 | 31.988 | 45.845 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 24.636 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 24.636 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 24.636 |
| 45.845 | 31.988 | 31.988 | -0.000 | -0.000 | -0.000 |
| 31.988 | 45.845 | 31.988 | -0.000 | -0.000 | -0.000 |
| 31.988 | 31.988 | 45.845 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 25.162 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 25.162 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 25.162 |
| 45.845 | 31.988 | 31.988 | 0.000 | 0.000 | 0.000 |
| 31.988 | 45.845 | 31.988 | 0.000 | 0.000 | -0.000 |
| 31.988 | 31.988 | 45.845 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 25.162 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 25.162 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 25.162 |
| 497.546 | 269.432 | 269.432 | -0.000 | 0.000 | 0.000 |
| 269.432 | 497.546 | 269.432 | 0.000 | 0.000 | -0.000 |
| 269.432 | 269.432 | 497.546 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 41.187 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 41.187 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 41.187 |
| 475.525 | 280.443 | 280.443 | 0.000 | -0.000 | 0.000 |
| 280.443 | 475.525 | 280.443 | 0.000 | 0.000 | -0.000 |
| 280.443 | 280.443 | 475.525 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 41.187 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 41.187 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 41.187 |
| 497.545 | 269.431 | 269.431 | -0.000 | 0.000 | -0.000 |
| 269.431 | 497.545 | 269.431 | 0.000 | 0.000 | -0.000 |
| 280.442 | 280.442 | 475.523 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -14.493 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -14.493 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -14.493 |
| 475.526 | 280.444 | 280.444 | 0.000 | -0.000 | 0.000 |
| 280.444 | 475.526 | 280.444 | 0.000 | -0.000 | 0.000 |
| 280.444 | 280.444 | 475.526 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -14.493 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -14.493 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -14.493 |
| 475.513 | 280.430 | 280.430 | 0.000 | 0.000 | -0.000 |
| 280.430 | 475.513 | 280.430 | 0.000 | 0.000 | 0.000 |
| 280.430 | 280.430 | 475.513 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.001 | 0.001 | -14.493 | 0.000 | 0.000 |
| 0.001 | 0.000 | 0.001 | -0.000 | -14.493 | -0.000 |
| 0.001 | 0.001 | 0.000 | -0.000 | -0.000 | -14.493 |
| 475.536 | 280.455 | 280.455 | 0.000 | 0.000 | 0.000 |
| 280.455 | 475.536 | 280.455 | 0.000 | -0.000 | 0.000 |
| 280.455 | 280.455 | 475.536 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.001 | -0.001 | -14.493 | -0.000 | 0.000 |
| -0.001 | -0.000 | -0.001 | 0.000 | -14.493 | -0.000 |
| -0.001 | -0.001 | -0.000 | 0.000 | 0.000 | -14.493 |
| 70.391 | 29.413 | 29.413 | 0.000 | -0.000 | 0.000 |
| 29.413 | 70.391 | 29.413 | 0.000 | 0.000 | 0.000 |
| 29.413 | 29.413 | 70.391 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 23.179 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 23.179 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 23.179 |
| 70.391 | 29.413 | 29.413 | -0.000 | -0.000 | 0.000 |
| 29.413 | 70.391 | 29.413 | 0.000 | 0.000 | 0.000 |
| 29.413 | 29.413 | 70.391 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 23.179 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 23.179 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 23.179 |
| 72.379 | 28.419 | 28.419 | 0.000 | 0.000 | 0.000 |
| 28.419 | 72.379 | 28.419 | -0.000 | 0.000 | 0.000 |
| 28.419 | 28.419 | 72.379 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 24.723 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 24.723 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 24.723 |
| 70.391 | 29.413 | 29.413 | -0.000 | 0.000 | -0.000 |
| 29.413 | 70.391 | 29.413 | -0.000 | 0.000 | -0.000 |
| 29.413 | 29.413 | 70.391 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 24.723 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 24.723 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 24.723 |
| 70.391 | 29.413 | 29.413 | -0.000 | -0.000 | 0.000 |
| 29.413 | 70.391 | 29.413 | -0.000 | -0.000 | 0.000 |
| 29.413 | 29.413 | 70.391 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 23.179 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 23.179 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 23.179 |
| 70.391 | 29.413 | 29.413 | -0.000 | 0.000 | 0.000 |
| 29.413 | 70.391 | 29.413 | 0.000 | 0.000 | -0.000 |
| 29.413 | 29.413 | 70.391 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 23.179 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 23.179 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 23.179 |
| 500.740 | 404.368 | 404.368 | 0.000 | -0.000 | -0.000 |
| 404.368 | 500.740 | 404.368 | -0.000 | -0.000 | 0.000 |
| 404.368 | 404.368 | 500.740 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 195.925 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 195.925 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 195.925 |
| 500.739 | 404.367 | 404.367 | 0.000 | 0.000 | 0.000 |
| 404.367 | 500.739 | 404.367 | 0.000 | 0.000 | 0.000 |
| 404.367 | 404.367 | 500.739 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 195.925 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 195.925 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 195.925 |
| 500.741 | 404.369 | 404.369 | 0.000 | -0.000 | -0.000 |
| 404.369 | 500.741 | 404.369 | -0.000 | -0.000 | 0.000 |
| 404.369 | 404.369 | 500.741 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 160.856 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 160.856 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 160.856 |
| 500.738 | 404.366 | 404.366 | -0.000 | 0.000 | 0.000 |
| 404.366 | 500.738 | 404.366 | 0.000 | 0.000 | -0.000 |
| 404.366 | 404.366 | 500.738 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 160.856 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 160.856 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 160.856 |
| 500.753 | 404.382 | 404.382 | 0.000 | -0.000 | 0.000 |
| 404.382 | 500.753 | 404.382 | 0.000 | -0.000 | 0.000 |
| 404.382 | 404.382 | 500.753 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.001 | -0.000 | 195.925 | 0.000 | 0.000 |
| -0.001 | 0.000 | -0.000 | 0.000 | 195.925 | 0.000 |
| -0.001 | -0.000 | 0.000 | 0.000 | 0.000 | 195.925 |
| 500.726 | 404.353 | 404.353 | -0.000 | 0.000 | -0.000 |
| 404.353 | 500.726 | 404.353 | -0.000 | 0.000 | -0.000 |
| 404.353 | 404.353 | 500.726 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.001 | 0.000 | 195.925 | -0.000 | -0.000 |
| 0.001 | -0.000 | 0.000 | -0.000 | 195.925 | -0.000 |
| 0.001 | 0.000 | -0.000 | -0.000 | -0.000 | 195.925 |
| 42.800 | 25.004 | 25.004 | 0.000 | -0.000 | -0.000 |
| 25.004 | 42.800 | 25.004 | -0.000 | 0.000 | -0.000 |
| 25.004 | 25.004 | 42.800 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 24.294 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 24.294 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 24.294 |
| 42.800 | 25.004 | 25.004 | 0.000 | -0.000 | 0.000 |
| 25.004 | 42.800 | 25.004 | 0.000 | 0.000 | 0.000 |
| 25.004 | 25.004 | 42.800 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 24.294 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 24.294 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 24.294 |
| 42.800 | 25.004 | 25.004 | 0.000 | -0.000 | -0.000 |
| 25.004 | 42.800 | 25.004 | -0.000 | -0.000 | -0.000 |
| 25.004 | 25.004 | 42.800 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 22.333 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 22.333 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 22.333 |
| 42.800 | 25.004 | 25.004 | -0.000 | -0.000 | 0.000 |
| 25.004 | 42.800 | 25.004 | 0.000 | 0.000 | 0.000 |
| 25.004 | 25.004 | 42.800 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 22.333 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 22.333 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 22.333 |
| 42.800 | 25.004 | 25.004 | -0.000 | -0.000 | -0.000 |
| 25.004 | 42.800 | 25.004 | -0.000 | 0.000 | -0.000 |
| 25.004 | 25.004 | 42.800 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 24.294 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 24.294 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 24.294 |
| 42.799 | 25.004 | 25.004 | -0.000 | -0.000 | 0.000 |
| 25.004 | 42.799 | 25.004 | 0.000 | -0.000 | 0.000 |
| 25.004 | 25.004 | 42.799 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 24.294 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 24.294 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 24.294 |
| 625.943 | 541.320 | 541.320 | 0.000 | 0.000 | 0.000 |
| 541.320 | 625.943 | 541.320 | 0.000 | 0.000 | -0.000 |
| 541.320 | 541.320 | 625.943 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 230.069 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 230.069 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 230.069 |
| 625.943 | 541.321 | 541.321 | -0.000 | -0.000 | -0.000 |
| 541.321 | 625.943 | 541.321 | -0.000 | 0.000 | 0.000 |
| 541.321 | 541.321 | 625.943 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 230.069 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 230.069 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 230.069 |
| 625.940 | 541.319 | 541.319 | 0.000 | -0.000 | 0.000 |
| 541.319 | 625.940 | 541.319 | 0.000 | 0.000 | 0.000 |
| 541.319 | 541.319 | 625.940 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 230.069 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 230.069 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 230.069 |
| 625.946 | 541.322 | 541.322 | -0.000 | -0.000 | 0.000 |
| 541.322 | 625.946 | 541.322 | -0.000 | 0.000 | -0.000 |
| 541.322 | 541.322 | 625.946 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 230.069 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 230.069 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 230.069 |
| 625.916 | 541.304 | 541.304 | 0.000 | -0.000 | 0.000 |
| 541.304 | 625.916 | 541.304 | 0.000 | -0.000 | -0.000 |
| 541.304 | 541.304 | 625.916 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.001 | 0.001 | 230.069 | -0.000 | -0.000 |
| 0.001 | 0.000 | 0.001 | 0.000 | 230.069 | 0.000 |
| 0.001 | 0.001 | 0.000 | 0.000 | 0.000 | 230.069 |
| 625.969 | 541.337 | 541.337 | -0.000 | 0.000 | 0.000 |
| 541.337 | 625.969 | 541.337 | -0.000 | -0.000 | -0.000 |
| 541.337 | 541.337 | 625.969 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.001 | -0.001 | 230.069 | -0.000 | 0.000 |
| -0.001 | -0.000 | -0.001 | -0.000 | 230.069 | -0.000 |
| -0.001 | -0.001 | -0.000 | -0.000 | -0.000 | 230.069 |
| 52.720 | 26.943 | 26.943 | 0.000 | -0.000 | -0.000 |
| 26.943 | 52.720 | 26.943 | -0.000 | -0.000 | 0.000 |
| 26.943 | 26.943 | 52.720 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 26.554 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 26.554 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 26.554 |
| 52.720 | 26.943 | 26.943 | -0.000 | 0.000 | 0.000 |
| 26.943 | 52.720 | 26.943 | 0.000 | 0.000 | 0.000 |
| 26.943 | 26.943 | 52.720 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 26.554 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 26.554 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 26.554 |
| 52.720 | 26.943 | 26.943 | 0.000 | -0.000 | -0.000 |
| 26.943 | 52.720 | 26.943 | 0.000 | -0.000 | -0.000 |
| 26.943 | 26.943 | 52.720 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 26.554 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 26.554 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 26.554 |
| 52.720 | 26.943 | 26.943 | -0.000 | 0.000 | 0.000 |
| 26.943 | 52.720 | 26.943 | -0.000 | 0.000 | 0.000 |
| 26.943 | 26.943 | 52.720 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 26.554 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 26.554 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 26.554 |
| 52.720 | 26.943 | 26.943 | 0.000 | -0.000 | -0.000 |
| 26.943 | 52.720 | 26.943 | -0.000 | -0.000 | -0.000 |
| 26.943 | 26.943 | 52.720 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 26.554 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 26.554 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 26.554 |
| 52.720 | 26.943 | 26.943 | -0.000 | 0.000 | -0.000 |
| 26.943 | 52.720 | 26.943 | 0.000 | 0.000 | 0.000 |
| 26.943 | 26.943 | 52.720 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 26.554 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 26.554 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 26.554 |
| 455.944 | 597.462 | 83.828 | 0.000 | -0.000 | -0.000 |
| 597.462 | 455.944 | 83.828 | -0.000 | 0.000 | 0.000 |
| 83.828 | 83.828 | -314.438 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 43.543 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 43.543 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 370.658 |
| 336.773 | 716.633 | 83.828 | 0.000 | 0.000 | -0.000 |
| 716.633 | 336.773 | 83.828 | 0.000 | 0.000 | -0.000 |
| 83.828 | 83.828 | -314.438 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 43.543 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 43.543 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 370.658 |
| 455.943 | 597.461 | 83.828 | -0.000 | -0.000 | 0.000 |
| 597.461 | 455.943 | 83.828 | 0.000 | 0.000 | -0.000 |
| 83.828 | 83.828 | -314.439 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 36.965 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 36.965 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 370.658 |
| 167590817.084 | 177299922.133 | 171252771.212 | 339815.619 | 1373997.758 | 760315.962 |
| 181403079.763 | 172972206.852 | 173930880.290 | 65594.346 | 835150.756 | -1643125.210 |
| 83.828 | 83.828 | -314.438 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 36.965 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 36.965 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 370.658 |
| -19675367.685 | -18609359.990 | -19546718.434 | -60270.450 | 98603.037 | 104.069 |
| -18971257.325 | -19697624.652 | -19738725.910 | -47358.894 | -76229.358 | -195438.196 |
| 83.828 | 83.828 | -314.441 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 36.965 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 36.965 | 0.000 |
| -0.001 | 0.000 | -0.000 | 0.000 | -0.000 | 370.658 |
| 18240134.910 | 19296373.158 | 18971674.985 | -33398.507 | 14560.767 | 56370.005 |
| 19718508.815 | 18492582.840 | 19819037.289 | -21397.635 | 146610.710 | 303935.562 |
| 83.829 | 83.829 | -314.435 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 36.965 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 36.965 | -0.000 |
| 0.001 | -0.000 | 0.000 | 0.000 | 0.000 | 370.658 |
| 169.271 | 202.226 | 202.226 | 0.000 | -0.000 | -0.000 |
| 202.226 | 169.271 | 202.226 | 0.000 | 0.000 | -0.000 |
| 202.226 | 202.226 | 169.271 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 72.771 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 72.771 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 72.771 |
| 169.271 | 202.227 | 202.226 | -0.000 | 0.000 | 0.000 |
| 202.227 | 169.271 | 202.226 | -0.000 | 0.000 | 0.000 |
| 202.227 | 202.227 | 169.271 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 142.606 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 142.606 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 72.771 |
| 169.271 | 202.226 | 202.226 | 0.000 | -0.000 | -0.000 |
| 202.226 | 169.271 | 202.226 | 0.000 | -0.000 | -0.000 |
| 202.226 | 202.226 | 169.271 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 142.606 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 142.606 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 142.606 |
| 169.271 | 202.227 | 202.227 | 0.000 | 0.000 | 0.000 |
| 202.227 | 169.271 | 202.227 | -0.000 | 0.000 | 0.000 |
| 202.227 | 202.227 | 169.271 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 142.606 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 142.606 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 142.606 |
| 169.270 | 202.224 | 202.224 | 0.000 | -0.000 | -0.000 |
| 202.224 | 169.270 | 202.224 | 0.000 | 0.000 | -0.000 |
| 202.224 | 202.224 | 169.270 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 72.771 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 72.771 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 72.771 |
| 169.272 | 202.229 | 202.229 | 0.000 | 0.000 | 0.000 |
| 202.229 | 169.272 | 202.229 | 0.000 | 0.000 | 0.000 |
| 202.229 | 202.229 | 169.272 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 72.771 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 72.771 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 72.771 |
| 282.839 | 205.436 | 205.436 | 0.000 | 0.000 | 0.000 |
| 205.436 | 282.839 | 205.436 | 0.000 | 0.000 | 0.000 |
| 205.436 | 205.436 | 282.839 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 91.152 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 91.152 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 91.152 |
| 282.839 | 205.436 | 205.436 | -0.000 | -0.000 | -0.000 |
| 205.436 | 282.839 | 205.436 | -0.000 | -0.000 | -0.000 |
| 205.436 | 205.436 | 282.839 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 91.152 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 91.152 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 91.152 |
| 282.839 | 205.436 | 205.436 | 0.000 | 0.000 | 0.000 |
| 205.436 | 282.839 | 205.436 | 0.000 | 0.000 | 0.000 |
| 205.436 | 205.436 | 282.839 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 144.598 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 144.598 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 144.598 |
| 282.839 | 205.436 | 205.436 | -0.000 | -0.000 | -0.000 |
| 205.436 | 282.839 | 205.436 | -0.000 | -0.000 | -0.000 |
| 205.436 | 205.436 | 282.839 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 144.598 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 144.598 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 144.598 |
| 282.841 | 205.438 | 205.438 | 0.000 | 0.000 | 0.000 |
| 205.438 | 282.841 | 205.438 | 0.000 | -0.000 | 0.000 |
| 205.438 | 205.438 | 282.841 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 144.598 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 144.598 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 144.598 |
| 282.837 | 205.435 | 205.435 | -0.000 | -0.000 | -0.000 |
| 205.435 | 282.837 | 205.435 | -0.000 | -0.000 | -0.000 |
| 205.435 | 205.435 | 282.837 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 144.598 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 144.598 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 144.598 |
| 318.927 | 206.527 | 206.527 | -0.000 | -0.000 | -0.000 |
| 206.527 | 318.927 | 206.527 | -0.000 | -0.000 | -0.000 |
| 206.527 | 206.527 | 318.927 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 115.668 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 115.668 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 115.668 |
| 318.927 | 206.527 | 206.527 | 0.000 | 0.000 | 0.000 |
| 206.527 | 318.927 | 206.527 | 0.000 | 0.000 | 0.000 |
| 206.527 | 206.527 | 318.927 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 115.668 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 115.668 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 115.668 |
| 318.927 | 206.527 | 206.527 | -0.000 | -0.000 | -0.000 |
| 206.527 | 318.927 | 206.527 | -0.000 | 0.000 | -0.000 |
| 206.527 | 206.527 | 318.927 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 115.668 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 115.668 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 115.668 |
| 318.927 | 206.527 | 206.527 | 0.000 | 0.000 | 0.000 |
| 206.527 | 318.927 | 206.527 | 0.000 | 0.000 | -0.000 |
| 206.527 | 206.527 | 318.927 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 115.668 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 115.668 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 115.668 |
| 318.925 | 206.526 | 206.526 | -0.000 | 0.000 | -0.000 |
| 206.526 | 318.925 | 206.526 | -0.000 | -0.000 | 0.000 |
| 206.526 | 206.526 | 318.925 | -0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 115.668 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 115.668 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 115.668 |
| 318.929 | 206.528 | 206.528 | 0.000 | -0.000 | -0.000 |
| 206.528 | 318.929 | 206.528 | 0.000 | 0.000 | -0.000 |
| 206.528 | 206.528 | 318.929 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 115.668 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 115.668 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 115.668 |
| 278.686 | 359.054 | 359.054 | -0.000 | -0.000 | -0.000 |
| 359.054 | 278.686 | 359.054 | -0.000 | -0.000 | -0.000 |
| 359.054 | 359.054 | 278.686 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 146.050 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 146.050 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 146.050 |
| 278.685 | 359.054 | 359.054 | 0.000 | 0.000 | 0.000 |
| 359.054 | 278.685 | 359.054 | 0.000 | 0.000 | 0.000 |
| 359.054 | 359.054 | 278.685 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 146.050 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 146.050 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 146.050 |
| 278.686 | 359.055 | 359.055 | -0.000 | -0.000 | -0.000 |
| 359.055 | 278.686 | 359.055 | -0.000 | -0.000 | -0.000 |
| 359.055 | 359.055 | 278.686 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 146.050 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 146.050 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 146.050 |
| 278.685 | 359.053 | 359.053 | 0.000 | 0.000 | 0.000 |
| 359.053 | 278.685 | 359.053 | 0.000 | 0.000 | 0.000 |
| 359.053 | 359.053 | 278.685 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 146.050 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 146.050 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 146.050 |
| 278.692 | 359.063 | 359.063 | -0.000 | -0.000 | -0.000 |
| 359.063 | 278.692 | 359.063 | -0.000 | -0.000 | -0.000 |
| 359.063 | 359.063 | 278.692 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 164.985 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 164.985 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 164.985 |
| 278.679 | 359.046 | 359.046 | 0.000 | 0.000 | 0.000 |
| 359.046 | 278.679 | 359.046 | 0.000 | 0.000 | 0.000 |
| 359.046 | 359.046 | 278.679 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 164.985 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 164.985 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 164.985 |
| 23.829 | 16.936 | 16.936 | -0.000 | -0.000 | -0.000 |
| 16.936 | 23.829 | 16.936 | -0.000 | -0.000 | -0.000 |
| 16.936 | 16.936 | 23.829 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 16.430 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 16.430 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 16.430 |
| 23.829 | 16.936 | 16.936 | -0.000 | 0.000 | 0.000 |
| 16.936 | 23.829 | 16.936 | 0.000 | 0.000 | 0.000 |
| 16.936 | 16.936 | 23.829 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 16.430 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 16.430 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 16.430 |
| 23.829 | 16.936 | 16.936 | -0.000 | -0.000 | -0.000 |
| 16.936 | 23.829 | 16.936 | -0.000 | -0.000 | -0.000 |
| 16.936 | 16.936 | 23.829 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 16.430 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 16.430 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 16.430 |
| 23.829 | 16.936 | 16.936 | -0.000 | 0.000 | 0.000 |
| 16.936 | 23.829 | 16.936 | 0.000 | 0.000 | 0.000 |
| 16.936 | 16.936 | 23.829 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 16.430 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 16.430 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 16.430 |
| 23.829 | 16.936 | 16.936 | -0.000 | -0.000 | -0.000 |
| 16.936 | 23.829 | 16.936 | -0.000 | -0.000 | -0.000 |
| 16.936 | 16.936 | 23.829 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 12.345 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 12.345 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 12.345 |
| 23.829 | 16.936 | 16.936 | 0.000 | 0.000 | 0.000 |
| 16.936 | 23.829 | 16.936 | 0.000 | 0.000 | 0.000 |
| 16.936 | 16.936 | 23.829 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 12.345 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 12.345 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 12.345 |
| 173.440 | 52.749 | 52.749 | 0.000 | 0.000 | 0.000 |
| 52.749 | 173.440 | 52.749 | 0.000 | 0.000 | 0.000 |
| 52.749 | 52.749 | 173.440 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 25.514 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 25.514 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 25.514 |
| 173.440 | 52.749 | 52.749 | -0.000 | -0.000 | -0.000 |
| 52.749 | 173.440 | 52.749 | -0.000 | -0.000 | -0.000 |
| 52.749 | 52.749 | 173.440 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 25.514 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 25.514 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 25.514 |
| 173.440 | 52.748 | 52.748 | 0.000 | 0.000 | 0.000 |
| 52.748 | 173.440 | 52.748 | 0.000 | 0.000 | 0.000 |
| 52.748 | 52.748 | 173.440 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 25.514 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 25.514 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 25.514 |
| 173.440 | 52.749 | 52.749 | -0.000 | -0.000 | -0.000 |
| 52.749 | 173.440 | 52.749 | -0.000 | -0.000 | -0.000 |
| 52.749 | 52.749 | 173.440 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 25.514 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 25.514 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 25.514 |
| 173.441 | 52.748 | 52.748 | 0.000 | 0.000 | 0.000 |
| 52.748 | 173.441 | 52.748 | 0.000 | 0.000 | 0.000 |
| 52.748 | 52.748 | 173.441 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 25.514 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 25.514 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 25.514 |
| 173.440 | 52.749 | 52.749 | -0.000 | 0.000 | -0.000 |
| 52.749 | 173.440 | 52.749 | -0.000 | -0.000 | -0.000 |
| 52.749 | 52.749 | 173.440 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 25.514 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 25.514 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 25.514 |
| 192.772 | 187.961 | 187.961 | -0.000 | 0.000 | -0.000 |
| 187.961 | 192.772 | 187.961 | 0.000 | -0.000 | -0.000 |
| 187.961 | 187.961 | 192.772 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 83.490 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 83.490 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 83.490 |
| 192.772 | 187.961 | 187.961 | 0.000 | 0.000 | 0.000 |
| 187.961 | 192.772 | 187.961 | 0.000 | 0.000 | 0.000 |
| 187.961 | 187.961 | 192.772 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 83.490 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 83.490 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 83.490 |
| 192.772 | 187.960 | 187.960 | -0.000 | -0.000 | -0.000 |
| 187.960 | 192.772 | 187.960 | -0.000 | -0.000 | -0.000 |
| 187.960 | 187.960 | 192.772 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 83.490 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 83.490 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 83.490 |
| 192.773 | 187.961 | 187.961 | 0.000 | 0.000 | 0.000 |
| 187.961 | 192.773 | 187.961 | 0.000 | 0.000 | 0.000 |
| 187.961 | 187.961 | 192.773 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 83.490 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 83.490 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 83.490 |
| 192.766 | 187.954 | 187.954 | -0.000 | -0.000 | -0.000 |
| 187.954 | 192.766 | 187.954 | -0.000 | -0.000 | -0.000 |
| 187.954 | 187.954 | 192.766 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 83.490 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 83.490 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 83.490 |
| 192.778 | 187.967 | 187.967 | 0.000 | 0.000 | 0.000 |
| 187.967 | 192.778 | 187.967 | 0.000 | 0.000 | 0.000 |
| 187.967 | 187.967 | 192.778 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 83.490 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 83.490 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 83.490 |
| 42.894 | -0.133 | -0.133 | -0.000 | 0.000 | 0.000 |
| -0.133 | 42.894 | -0.133 | 0.000 | -0.000 | -0.000 |
| -0.133 | -0.133 | 42.894 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.491 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.491 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -0.491 |
| 42.894 | -0.133 | -0.133 | 0.000 | 0.000 | 0.000 |
| -0.133 | 42.894 | -0.133 | 0.000 | 0.000 | 0.000 |
| -0.133 | -0.133 | 42.894 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.491 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.491 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -0.491 |
| 42.894 | -0.133 | -0.133 | -0.000 | -0.000 | -0.000 |
| -0.133 | 42.894 | -0.133 | -0.000 | -0.000 | -0.000 |
| -0.133 | -0.133 | 42.894 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.491 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.491 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -0.491 |
| 42.894 | -0.133 | -0.133 | -0.000 | 0.000 | 0.000 |
| -0.133 | 42.894 | -0.133 | 0.000 | -0.000 | 0.000 |
| -0.133 | -0.133 | 42.894 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.491 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.491 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -0.491 |
| 42.894 | -0.133 | -0.133 | -0.000 | -0.000 | -0.000 |
| -0.133 | 42.894 | -0.133 | -0.000 | -0.000 | 0.000 |
| -0.133 | -0.133 | 42.894 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.491 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.491 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -0.491 |
| 42.894 | -0.133 | -0.133 | 0.000 | 0.000 | 0.000 |
| -0.133 | 42.894 | -0.133 | 0.000 | 0.000 | 0.000 |
| -0.133 | -0.133 | 42.894 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.491 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.491 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -0.491 |
| 55.319 | 60.335 | 60.335 | 0.000 | 0.000 | 0.000 |
| 60.335 | 55.319 | 60.335 | 0.000 | 0.000 | -0.000 |
| 60.335 | 60.335 | 55.319 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 49.103 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 49.103 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 49.103 |
| 55.319 | 60.335 | 60.335 | -0.000 | -0.000 | -0.000 |
| 60.335 | 55.319 | 60.335 | -0.000 | -0.000 | 0.000 |
| 60.335 | 60.335 | 55.319 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 49.103 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 49.103 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 49.103 |
| 55.319 | 60.335 | 60.335 | -0.000 | 0.000 | -0.000 |
| 60.335 | 55.319 | 60.335 | 0.000 | -0.000 | -0.000 |
| 60.335 | 60.335 | 55.319 | -0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 15.293 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 15.293 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 15.293 |
| 55.319 | 60.335 | 60.335 | -0.000 | -0.000 | -0.000 |
| 60.335 | 55.319 | 60.335 | -0.000 | -0.000 | 0.000 |
| 60.335 | 60.335 | 55.319 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 15.293 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 15.293 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 15.293 |
| 55.319 | 60.335 | 60.335 | -0.000 | 0.000 | -0.000 |
| 60.335 | 55.319 | 60.335 | 0.000 | -0.000 | -0.000 |
| 60.335 | 60.335 | 55.319 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 15.293 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 15.293 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 15.293 |
| 55.319 | 60.335 | 60.335 | -0.000 | -0.000 | -0.000 |
| 60.335 | 55.319 | 60.335 | -0.000 | -0.000 | 0.000 |
| 60.335 | 60.335 | 55.319 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 15.293 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 15.293 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 15.293 |
| 29.784 | 18.838 | 18.838 | 0.000 | 0.000 | -0.000 |
| 18.838 | 29.784 | 18.838 | 0.000 | 0.000 | 0.000 |
| 18.838 | 18.838 | 29.784 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 11.491 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 11.491 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 11.491 |
| 29.784 | 18.838 | 18.838 | -0.000 | -0.000 | -0.000 |
| 18.838 | 29.784 | 18.838 | -0.000 | -0.000 | -0.000 |
| 18.838 | 18.838 | 29.784 | -0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 11.491 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 11.491 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 11.491 |
| 29.784 | 18.838 | 18.838 | 0.000 | -0.000 | -0.000 |
| 18.838 | 29.784 | 18.838 | 0.000 | 0.000 | 0.000 |
| 18.838 | 18.838 | 29.784 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 11.491 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 11.491 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 11.491 |
| 29.784 | 18.838 | 18.838 | -0.000 | -0.000 | -0.000 |
| 18.838 | 29.784 | 18.838 | -0.000 | -0.000 | -0.000 |
| 18.838 | 18.838 | 29.784 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 11.491 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 11.491 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 11.491 |
| 29.784 | 18.838 | 18.838 | 0.000 | 0.000 | -0.000 |
| 18.838 | 29.784 | 18.838 | 0.000 | 0.000 | 0.000 |
| 18.838 | 18.838 | 29.784 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 11.491 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 11.491 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 11.491 |
| 29.785 | 18.838 | 18.838 | -0.000 | -0.000 | -0.000 |
| 18.838 | 29.785 | 18.838 | -0.000 | -0.000 | -0.000 |
| 18.838 | 18.838 | 29.785 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 11.491 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 11.491 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 11.491 |
| -606.711 | -717.591 | -96.711 | 13.060 | 12.663 | -0.000 |
| -717.591 | -606.711 | -96.711 | -13.060 | -12.663 | -0.000 |
| -736.006 | -736.006 | 59.392 | 0.000 | 0.000 | -0.000 |
| -2559.791 | -2582.693 | -349.757 | 37.260 | 0.000 | -11.606 |
| -842.244 | -867.569 | -214.932 | 0.000 | 39.956 | 13.060 |
| -854.907 | -854.907 | -214.932 | -12.663 | 13.060 | 55.442 |
| 256.600 | 136.797 | 119.223 | 13.472 | 12.818 | 0.000 |
| 136.797 | 256.600 | 119.223 | -13.472 | -12.818 | 0.000 |
| 119.635 | 119.635 | 275.571 | -0.000 | -0.000 | 0.000 |
| 2582.581 | 2559.680 | 349.750 | 37.260 | -0.000 | -11.606 |
| 2582.736 | 2559.524 | 349.750 | -0.000 | 37.260 | 11.451 |
| 854.907 | 854.907 | 214.932 | -12.663 | 13.060 | 55.442 |
| 256.600 | 136.796 | 119.223 | 13.472 | 12.818 | -0.000 |
| 51.837 | 162.667 | 96.725 | -13.058 | -12.662 | -0.000 |
| 33.410 | 33.410 | 252.833 | 0.000 | 0.000 | -0.000 |
| -72.427 | -98.547 | -21.492 | 39.956 | 0.000 | -12.663 |
| -72.824 | -98.150 | -21.492 | 0.000 | 39.956 | 13.060 |
| -85.491 | -85.491 | -21.493 | -12.663 | 13.060 | 55.442 |
| 256.601 | 136.797 | 119.224 | 13.472 | 12.818 | 0.000 |
| 398.355 | 498.151 | 152.154 | -11.443 | -11.614 | 0.000 |
| 119.635 | 119.635 | 275.571 | -0.000 | -0.000 | 0.000 |
| 13.472 | -13.472 | 0.000 | 44.970 | -0.000 | -12.818 |
| 243.859 | 221.625 | 22.959 | -0.000 | 36.458 | 12.533 |
| 0.000 | 0.000 | -0.000 | -12.818 | 13.472 | 59.902 |
| 239.327 | 128.903 | 116.048 | 13.039 | 12.655 | -0.000 |
| 136.795 | 256.599 | 119.222 | -13.473 | -12.818 | -0.000 |
| 110.358 | 110.358 | 272.193 | 0.000 | 0.000 | -0.000 |
| 13.472 | -13.472 | -0.000 | 44.970 | 0.000 | -12.818 |
| 12.818 | -12.818 | -0.000 | 0.000 | 44.970 | 13.472 |
| -0.000 | -0.000 | 0.000 | -12.818 | 13.472 | 59.902 |
| 257.231 | 145.769 | 120.421 | 13.087 | 12.673 | 0.000 |
| 145.769 | 257.231 | 120.421 | -13.087 | -12.673 | 0.000 |
| 127.439 | 127.439 | 276.454 | -0.000 | -0.000 | 0.000 |
| 35.328 | 10.222 | 2.263 | 36.617 | -0.000 | -11.159 |
| 39.447 | 16.474 | 3.611 | -0.000 | 38.459 | 11.474 |
| 0.000 | 0.000 | -0.000 | -12.818 | 13.472 | 59.902 |
| -1239.855 | -1352.070 | -833.497 | 12.451 | -12.074 | -0.000 |
| -2899.629 | -2792.366 | -1225.044 | -12.155 | 11.210 | 0.000 |
| -1371.863 | -1371.863 | -674.451 | -0.000 | 0.000 | 0.000 |
| 12.884 | -12.884 | 0.000 | 44.334 | 0.000 | 12.216 |
| -12.216 | 12.216 | -0.000 | 0.000 | 44.334 | 12.884 |
| -3042.505 | -3042.504 | -1346.171 | 11.210 | 12.155 | 53.632 |
| 1747.864 | 1635.644 | 1076.800 | 12.451 | -12.074 | 0.000 |
| 3185.379 | 3292.645 | 1467.297 | -12.155 | 11.210 | -0.000 |
| 3164.376 | 3164.376 | 1626.431 | -0.000 | -0.000 | 0.000 |
| 3054.659 | 3030.348 | 1346.170 | 37.271 | 0.000 | 11.210 |
| 1481.784 | 1505.931 | 955.149 | 0.000 | 39.448 | 12.451 |
| -0.000 | 0.000 | 0.000 | 12.216 | 12.884 | 60.468 |
| 261.876 | 140.941 | 122.581 | 12.884 | -12.216 | 0.000 |
| -362.521 | -258.432 | -27.840 | -11.291 | 10.681 | -0.000 |
| -182.379 | -182.379 | 145.646 | -0.000 | 0.000 | -0.000 |
| -136.933 | -161.835 | -95.514 | 39.448 | 0.000 | 12.074 |
| -161.458 | -137.310 | -95.514 | 0.000 | 39.448 | 12.451 |
| -0.000 | -0.000 | 0.000 | 12.216 | 12.884 | 60.468 |
| 403.409 | 291.167 | 217.167 | 12.453 | -12.074 | -0.000 |
| 651.270 | 755.345 | 269.963 | -11.297 | 10.685 | -0.000 |
| 122.582 | 122.582 | 281.644 | 0.000 | 0.000 | 0.000 |
| 161.835 | 136.933 | 95.514 | 39.448 | 0.000 | 12.074 |
| 293.033 | 315.452 | 134.613 | -0.000 | 37.271 | 12.156 |
| 304.250 | 304.250 | 134.616 | 11.210 | 12.156 | 53.632 |
| 238.894 | 126.910 | 112.099 | 12.440 | -12.070 | 0.000 |
| 112.536 | 219.633 | 107.684 | -12.147 | 11.201 | 0.000 |
| 107.074 | 107.074 | 271.162 | -0.000 | -0.000 | -0.000 |
| -18.188 | -42.503 | -13.424 | 37.290 | 0.000 | 11.212 |
| -26.995 | -2.847 | -9.540 | -0.000 | 39.454 | 12.452 |
| -0.000 | -0.000 | 0.000 | 12.216 | 12.884 | 60.467 |
| 261.878 | 140.942 | 122.583 | 12.884 | -12.215 | -0.000 |
| 156.673 | 269.149 | 131.213 | -12.464 | 12.078 | -0.000 |
| 152.291 | 152.291 | 293.702 | 0.000 | 0.000 | 0.000 |
| 27.373 | 2.469 | 9.540 | 39.454 | 0.000 | 12.074 |
| 2.847 | 26.995 | 9.540 | -0.000 | 39.454 | 12.452 |
| 50.502 | 50.502 | 14.867 | 10.690 | 11.300 | 52.061 |
| 275.802 | 150.367 | 150.367 | 0.000 | 0.000 | 0.000 |
| 150.367 | 275.802 | 150.367 | 0.000 | -0.000 | 0.000 |
| 150.367 | 150.367 | 275.802 | 0.000 | 0.000 | 0.000 |
| 12.713 | 12.713 | 12.713 | 55.495 | -0.000 | -0.000 |
| 12.713 | 12.713 | 12.713 | -0.000 | 55.495 | 0.000 |
| 12.713 | 12.713 | 12.713 | 0.000 | -0.000 | 55.495 |
| 247.418 | 125.937 | 125.937 | 0.000 | 0.000 | 0.000 |
| 125.937 | 247.418 | 125.937 | 0.000 | 0.000 | 0.000 |
| 125.937 | 125.937 | 247.418 | 0.000 | 0.000 | 0.000 |
| -12.713 | -12.713 | -12.713 | 55.495 | 0.000 | 0.000 |
| -12.713 | -12.713 | -12.713 | -0.000 | 55.495 | 0.000 |
| -12.713 | -12.713 | -12.713 | 0.000 | 0.000 | 55.495 |
| 268.451 | 137.978 | 137.978 | -0.000 | -0.000 | -0.000 |
| 137.978 | 268.451 | 137.978 | -0.000 | -0.000 | -0.000 |
| 137.978 | 137.978 | 268.451 | -0.000 | -0.000 | 0.000 |
| 1.320 | 1.320 | 1.320 | 55.608 | 0.000 | 0.000 |
| 1.320 | 1.320 | 1.320 | 0.000 | 55.608 | 0.000 |
| 1.320 | 1.320 | 1.320 | 0.000 | 0.000 | 55.608 |
| 259.340 | 134.376 | 134.376 | -0.000 | -0.000 | -0.000 |
| 134.376 | 259.340 | 134.376 | -0.000 | -0.000 | -0.000 |
| 134.376 | 134.376 | 259.340 | -0.000 | -0.000 | -0.000 |
| -1.320 | -1.320 | -1.320 | 55.608 | -0.000 | -0.000 |
| -1.320 | -1.320 | -1.320 | -0.000 | 55.608 | -0.000 |
| -1.320 | -1.320 | -1.320 | -0.000 | -0.000 | 55.608 |
| 268.447 | 137.973 | 137.973 | 0.000 | -0.000 | -0.000 |
| 137.973 | 268.447 | 137.973 | -0.000 | -0.000 | -0.000 |
| 137.973 | 137.973 | 268.447 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 63.041 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 63.041 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 63.041 |
| 259.715 | 135.489 | 135.489 | -0.000 | -0.000 | -0.000 |
| 135.489 | 259.715 | 135.489 | -0.000 | -0.000 | -0.000 |
| 135.489 | 135.489 | 259.715 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 63.041 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 63.041 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 63.041 |
| 220.388 | 94.433 | 94.433 | 0.000 | 0.000 | 0.000 |
| 94.433 | 220.388 | 94.433 | 0.000 | 0.000 | 0.000 |
| 94.433 | 94.433 | 220.388 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 63.031 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 63.031 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 63.031 |
| 270.146 | 139.694 | 139.694 | -0.000 | -0.000 | -0.000 |
| 139.694 | 270.146 | 139.694 | -0.000 | -0.000 | -0.000 |
| 139.694 | 139.694 | 270.146 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 63.031 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 63.031 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 63.031 |
| 254.706 | 136.185 | 136.185 | 0.000 | 0.000 | 0.000 |
| 136.185 | 254.706 | 136.185 | 0.000 | 0.000 | 0.000 |
| 136.185 | 136.185 | 254.706 | -0.000 | 0.000 | 0.000 |
| -4.420 | -4.420 | -4.420 | 55.763 | 0.000 | 0.000 |
| -4.420 | -4.420 | -4.420 | 0.000 | 55.763 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 63.031 |
| 270.147 | 139.695 | 139.695 | -0.000 | -0.000 | -0.000 |
| 139.695 | 270.147 | 139.695 | -0.000 | -0.000 | -0.000 |
| 139.695 | 139.695 | 270.147 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 63.031 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 63.031 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 63.031 |
| 270.142 | 139.689 | 139.689 | 0.000 | 0.000 | 0.000 |
| 139.689 | 270.142 | 139.689 | 0.000 | 0.000 | 0.000 |
| 139.689 | 139.689 | 270.142 | 0.000 | 0.000 | 0.000 |
| -0.544 | -0.544 | -0.544 | 53.750 | 0.000 | 0.000 |
| -0.544 | -0.544 | -0.544 | 0.000 | 53.750 | 0.000 |
| -0.544 | -0.544 | -0.544 | 0.000 | 0.000 | 53.750 |
| 261.456 | 137.767 | 137.767 | -0.000 | -0.000 | -0.000 |
| 137.767 | 261.456 | 137.767 | -0.000 | -0.000 | -0.000 |
| 137.767 | 137.767 | 261.456 | -0.000 | -0.000 | -0.000 |
| 0.544 | 0.544 | 0.544 | 53.750 | -0.000 | -0.000 |
| 0.544 | 0.544 | 0.544 | -0.000 | 53.750 | -0.000 |
| 0.544 | 0.544 | 0.544 | -0.000 | -0.000 | 53.750 |
| 3574.679 | 3463.923 | 496.697 | 12.989 | -12.598 | 0.000 |
| 3463.923 | 3574.679 | 496.697 | -12.989 | 12.598 | -0.000 |
| 119.907 | 119.907 | 275.970 | -0.000 | 0.000 | -0.000 |
| 3338.306 | 3312.328 | 377.995 | 39.776 | 0.000 | 12.598 |
| 3614.924 | 3638.885 | 541.610 | 0.000 | 38.724 | 12.159 |
| 0.000 | -0.000 | -0.000 | 12.758 | 13.413 | 59.949 |
| -3075.954 | -3186.712 | -259.292 | 12.989 | -12.598 | 0.000 |
| -3486.976 | -3381.515 | -423.610 | -12.159 | 11.981 | 0.000 |
| -3206.356 | -3206.356 | -103.333 | 0.000 | -0.000 | 0.000 |
| -3312.328 | -3338.306 | -377.995 | 39.776 | 0.000 | 12.598 |
| -3337.915 | -3312.719 | -377.995 | -0.000 | 39.776 | 12.989 |
| -3626.905 | -3626.904 | -541.610 | 11.981 | 12.159 | 52.731 |
| 581.886 | 471.139 | 156.501 | 12.989 | -12.598 | 0.000 |
| 471.139 | 581.886 | 156.501 | -12.989 | 12.598 | -0.000 |
| 451.493 | 451.493 | 312.461 | 0.000 | -0.000 | -0.000 |
| 366.772 | 341.205 | 63.365 | 39.205 | 0.000 | 11.621 |
| 319.932 | 345.128 | 37.799 | -0.000 | 39.777 | 12.989 |
| -0.000 | -0.000 | 0.000 | 12.758 | 13.413 | 59.949 |
| -117.306 | -222.758 | 63.837 | 12.159 | -11.980 | -0.000 |
| 137.170 | 257.068 | 119.535 | -13.413 | 12.758 | -0.000 |
| 119.907 | 119.907 | 275.970 | -0.000 | -0.000 | 0.000 |
| -341.226 | -366.792 | -63.368 | 39.205 | 0.000 | 11.621 |
| -12.758 | 12.758 | 0.000 | -0.000 | 44.908 | 13.413 |
| -332.532 | -332.531 | -37.799 | 12.598 | 12.989 | 55.379 |
| 282.890 | 174.437 | 124.513 | 12.787 | -11.641 | 0.000 |
| 171.872 | 282.534 | 122.473 | -12.985 | 12.596 | 0.000 |
| 119.906 | 119.906 | 275.971 | 0.000 | -0.000 | -0.000 |
| 13.413 | -13.413 | -0.000 | 44.908 | -0.000 | 12.758 |
| -12.758 | 12.758 | -0.000 | 0.000 | 44.908 | 13.413 |
| -0.000 | -0.000 | 0.000 | 12.758 | 13.413 | 59.949 |
| 216.185 | 105.335 | 114.932 | 12.993 | -12.599 | -0.000 |
| 105.335 | 216.185 | 114.932 | -12.993 | 12.599 | -0.000 |
| 85.710 | 85.710 | 270.879 | 0.000 | -0.000 | 0.000 |
| -24.092 | -48.416 | -5.422 | 38.727 | 0.000 | 11.978 |
| -47.027 | -23.781 | -6.332 | -0.000 | 39.205 | 12.784 |
| -33.251 | -33.251 | -3.780 | 12.598 | 12.989 | 55.379 |
| 258.114 | 133.802 | 133.802 | 0.000 | 0.000 | 0.000 |
| 133.802 | 258.114 | 133.802 | 0.000 | 0.000 | 0.000 |
| 133.802 | 133.802 | 258.114 | 0.000 | 0.000 | 0.000 |
| -0.190 | -0.190 | -0.190 | 53.760 | 0.000 | 0.000 |
| -0.190 | -0.190 | -0.190 | 0.000 | 53.760 | 0.000 |
| -0.190 | -0.190 | -0.190 | 0.000 | 0.000 | 53.760 |
| 258.173 | 134.135 | 134.135 | -0.000 | -0.000 | -0.000 |
| 134.135 | 258.173 | 134.135 | -0.000 | -0.000 | -0.000 |
| 134.135 | 134.135 | 258.173 | -0.000 | -0.000 | -0.000 |
| 0.190 | 0.190 | 0.190 | 53.760 | -0.000 | -0.000 |
| 0.190 | 0.190 | 0.190 | -0.000 | 53.760 | -0.000 |
| 0.190 | 0.190 | 0.190 | -0.000 | -0.000 | 53.760 |
| 266.836 | 136.235 | 136.235 | 0.000 | 0.000 | 0.000 |
| 136.235 | 266.836 | 136.235 | 0.000 | 0.000 | 0.000 |
| 136.235 | 136.235 | 266.836 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 63.077 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 63.077 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 63.077 |
| 258.150 | 133.983 | 133.983 | -0.000 | -0.000 | -0.000 |
| 133.983 | 258.150 | 133.983 | -0.000 | -0.000 | -0.000 |
| 133.983 | 133.983 | 258.150 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 63.077 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 63.077 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 63.077 |
| 255.680 | 137.180 | 137.180 | 0.000 | 0.000 | 0.000 |
| 137.180 | 255.680 | 137.180 | 0.000 | 0.000 | 0.000 |
| 137.180 | 137.180 | 255.680 | 0.000 | 0.000 | 0.000 |
| -0.002 | -0.002 | -0.002 | 53.757 | 0.000 | 0.000 |
| -0.001 | -0.001 | -0.002 | 0.000 | 53.314 | 0.000 |
| -0.002 | -0.001 | -0.001 | 0.000 | 0.000 | 53.314 |
| 258.151 | 133.972 | 133.972 | -0.000 | -0.000 | -0.000 |
| 133.972 | 258.151 | 133.972 | -0.000 | -0.000 | -0.000 |
| 133.972 | 133.972 | 258.151 | -0.000 | -0.000 | -0.000 |
| 0.002 | 0.002 | 0.002 | 53.757 | -0.000 | -0.000 |
| 0.001 | 0.001 | 0.002 | -0.000 | 53.314 | -0.000 |
| 0.002 | 0.002 | 0.002 | -0.000 | -0.000 | 53.757 |
| 507.662 | 205.488 | 205.488 | -0.000 | -0.000 | 0.000 |
| 205.488 | 507.662 | 205.488 | -0.000 | -0.000 | -0.000 |
| 205.488 | 205.488 | 507.662 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 136.587 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 136.587 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 136.587 |
| 507.662 | 205.488 | 205.488 | 0.000 | 0.000 | 0.000 |
| 205.488 | 507.662 | 205.488 | 0.000 | 0.000 | 0.000 |
| 205.488 | 205.488 | 507.662 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 136.587 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 136.587 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 136.587 |
| 507.662 | 205.488 | 205.488 | -0.000 | -0.000 | -0.000 |
| 205.488 | 507.662 | 205.488 | -0.000 | -0.000 | -0.000 |
| 205.488 | 205.488 | 507.662 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 117.220 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 117.220 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 117.220 |
| 507.662 | 205.488 | 205.488 | 0.000 | 0.000 | -0.000 |
| 205.488 | 507.662 | 205.488 | 0.000 | 0.000 | 0.000 |
| 205.488 | 205.488 | 507.662 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 117.220 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 117.220 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 117.220 |
| 507.665 | 205.489 | 205.489 | -0.000 | -0.000 | -0.000 |
| 205.489 | 507.665 | 205.489 | -0.000 | -0.000 | -0.000 |
| 205.489 | 205.489 | 507.665 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 136.587 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 136.587 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 136.587 |
| 507.659 | 205.487 | 205.487 | 0.000 | 0.000 | 0.000 |
| 205.487 | 507.659 | 205.487 | 0.000 | 0.000 | -0.000 |
| 205.487 | 205.487 | 507.659 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 136.587 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 136.587 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 136.587 |
| 68.330 | 15.308 | 15.308 | 0.000 | 0.000 | -0.000 |
| 15.308 | 68.330 | 15.308 | 0.000 | 0.000 | 0.000 |
| 15.308 | 15.308 | 68.330 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 18.215 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 18.215 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 18.215 |
| 68.330 | 15.308 | 15.308 | -0.000 | -0.000 | 0.000 |
| 15.308 | 68.330 | 15.308 | -0.000 | -0.000 | -0.000 |
| 15.308 | 15.308 | 68.330 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 18.215 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 18.215 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 18.215 |
| 68.330 | 15.308 | 15.308 | 0.000 | 0.000 | -0.000 |
| 15.308 | 68.330 | 15.308 | 0.000 | 0.000 | 0.000 |
| 15.308 | 15.308 | 68.330 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 18.252 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 18.252 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 18.252 |
| 68.330 | 15.308 | 15.308 | -0.000 | -0.000 | 0.000 |
| 15.308 | 68.330 | 15.308 | -0.000 | -0.000 | -0.000 |
| 15.308 | 15.308 | 68.330 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 18.252 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 18.252 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 18.252 |
| 68.330 | 15.308 | 15.308 | 0.000 | 0.000 | 0.000 |
| 15.308 | 68.330 | 15.308 | 0.000 | 0.000 | 0.000 |
| 15.308 | 15.308 | 68.330 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 18.215 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 18.215 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 18.215 |
| 68.331 | 15.308 | 15.308 | -0.000 | -0.000 | -0.000 |
| 15.308 | 68.331 | 15.308 | -0.000 | -0.000 | -0.000 |
| 15.308 | 15.308 | 68.331 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 18.215 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 18.215 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 18.215 |
| 1271.804 | 170.224 | 1442.952 | 0.000 | 0.000 | -0.000 |
| 1219.974 | 558.261 | 1551.363 | 0.000 | 0.000 | -0.000 |
| 179.555 | 273.880 | 522.693 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 136.057 | 0.000 | 0.000 |
| 1035.047 | -15.799 | 1270.840 | 0.000 | 19.595 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 49.912 |
| -1756.105 | 208.027 | -3104.069 | -0.000 | 0.000 | -0.000 |
| 187.011 | 579.222 | 274.898 | -0.000 | 0.000 | -0.000 |
| 179.555 | 273.880 | 522.693 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 136.057 | 0.000 | 0.000 |
| -1035.047 | 15.799 | -1270.840 | -0.000 | 19.595 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 49.912 |
| 401.917 | 168.828 | 489.632 | 0.000 | -0.000 | 0.000 |
| 370.137 | 566.708 | 611.857 | 0.000 | -0.000 | 0.000 |
| 269.550 | 277.739 | 613.196 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 136.057 | 0.000 | 0.000 |
| 102.840 | -1.570 | 126.268 | 0.000 | 19.751 | -0.000 |
| 196.352 | -3.541 | 326.954 | 0.000 | -0.000 | 49.780 |
| 133.411 | 187.633 | 44.933 | -0.000 | -0.000 | 0.000 |
| -21.902 | 574.043 | -41.395 | -0.000 | 0.000 | -0.000 |
| -63.953 | 299.236 | 32.523 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 136.057 | -0.000 | -0.000 |
| -102.840 | 1.570 | -126.268 | -0.000 | 19.751 | 0.000 |
| -196.352 | 3.541 | -326.954 | -0.000 | 0.000 | 49.780 |
| 263.886 | 188.205 | 181.767 | 0.000 | -0.000 | 0.000 |
| 192.448 | 570.085 | 313.249 | -0.000 | -0.000 | 0.000 |
| 179.556 | 273.882 | 522.697 | 0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 136.057 | -0.000 | 0.000 |
| 8.090 | -0.123 | 9.932 | -0.000 | 24.881 | -0.000 |
| 10.294 | -0.157 | 12.640 | 0.000 | 0.000 | 49.751 |
| 226.883 | 186.295 | 158.881 | -0.000 | -0.000 | 0.000 |
| 175.034 | 574.431 | 268.028 | 0.000 | -0.000 | 0.000 |
| 150.234 | 281.598 | 458.901 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 136.057 | 0.000 | 0.000 |
| -8.090 | 0.123 | -9.932 | -0.000 | 24.881 | 0.000 |
| -10.294 | 0.157 | -12.640 | 0.000 | -0.000 | 49.751 |
| -61.782 | -227.303 | 468.342 | 0.000 | -0.000 | 0.000 |
| 171.650 | 564.127 | 252.481 | -0.000 | -0.000 | -0.000 |
| -174.294 | -138.272 | 768.812 | 0.000 | -0.000 | 0.000 |
| -329.635 | -397.649 | 314.314 | 124.997 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 45.704 | -0.000 |
| -329.633 | -397.646 | 314.312 | 0.000 | -0.000 | 51.652 |
| 291.040 | 171.650 | 163.309 | -0.000 | 0.000 | 0.000 |
| 638.042 | 1270.260 | -825.043 | 0.000 | 0.000 | 0.000 |
| 163.309 | 252.482 | 480.175 | 0.000 | 0.000 | -0.000 |
| 329.635 | 397.649 | -314.314 | 124.997 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 45.704 | 0.000 |
| 329.633 | 397.646 | -314.312 | -0.000 | 0.000 | 51.652 |
| 291.040 | 171.650 | 163.309 | -0.000 | -0.000 | -0.000 |
| 171.650 | 564.127 | 252.481 | 0.000 | -0.000 | -0.000 |
| 122.360 | 219.098 | 486.941 | 0.000 | -0.000 | 0.000 |
| -47.566 | -71.041 | 107.521 | 121.164 | -0.000 | -0.000 |
| -32.948 | -39.746 | 31.416 | -0.000 | 31.133 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 51.866 |
| 291.040 | 171.650 | 163.309 | -0.000 | 0.000 | -0.000 |
| 171.650 | 564.127 | 252.481 | -0.000 | 0.000 | 0.000 |
| 163.310 | 252.482 | 480.174 | -0.000 | 0.000 | -0.000 |
| 32.984 | 39.789 | -31.450 | 124.991 | 0.000 | -0.000 |
| 32.948 | 39.746 | -31.416 | -0.000 | 31.133 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 51.866 |
| 249.002 | 155.985 | 159.018 | 0.000 | -0.000 | -0.000 |
| 158.087 | 550.380 | 267.736 | 0.000 | -0.000 | -0.000 |
| 163.308 | 252.481 | 480.176 | -0.000 | -0.000 | 0.000 |
| -3.424 | -4.131 | 3.266 | 124.663 | -0.000 | 0.000 |
| -3.223 | -3.888 | 3.073 | 0.000 | 31.450 | 0.000 |
| -3.284 | -3.961 | 3.132 | 0.000 | -0.000 | 51.652 |
| 253.780 | 168.509 | 136.282 | 0.000 | 0.000 | 0.000 |
| 171.651 | 564.127 | 252.482 | 0.000 | 0.000 | -0.000 |
| 150.878 | 270.662 | 373.015 | -0.000 | 0.000 | 0.000 |
| 3.424 | 4.131 | -3.266 | 124.663 | 0.000 | -0.000 |
| 3.223 | 3.888 | -3.073 | 0.000 | 31.450 | -0.000 |
| 4.576 | 6.671 | -9.597 | -0.000 | 0.000 | 51.707 |
| 4415.805 | -251.222 | 3963.962 | 0.000 | -0.000 | -0.000 |
| 4352.128 | 138.474 | 4067.363 | 0.000 | -0.000 | -0.000 |
| 4335.983 | -158.208 | 4280.046 | 0.000 | -0.000 | -0.000 |
| 9128.802 | 177.359 | 14488.673 | 122.363 | -0.000 | -0.000 |
| 4179.549 | -429.538 | 3799.969 | 0.000 | 25.465 | -0.000 |
| 9424.598 | 1014.496 | 10918.230 | 0.000 | 0.000 | 48.976 |
| -3943.342 | 607.856 | -3636.013 | -0.000 | 0.000 | 0.000 |
| -9247.882 | -450.836 | -10627.246 | -0.000 | -0.000 | -0.000 |
| -4023.250 | 700.894 | -3320.077 | -0.000 | 0.000 | 0.000 |
| -9128.802 | -177.359 | -14488.673 | 122.363 | 0.000 | 0.000 |
| -4179.549 | 429.538 | -3799.969 | -0.000 | 25.465 | 0.000 |
| -4179.559 | 429.539 | -3799.979 | -0.000 | 0.000 | 49.016 |
| 1130.772 | 191.365 | 1583.881 | 0.000 | -0.000 | -0.000 |
| 178.945 | 571.845 | 267.133 | 0.000 | 0.000 | 0.000 |
| 574.693 | 228.294 | 860.614 | 0.000 | -0.000 | -0.000 |
| 417.974 | -42.956 | 380.015 | 126.688 | -0.000 | -0.000 |
| 417.853 | -42.943 | 379.904 | 0.000 | 25.470 | -0.000 |
| 417.955 | -42.954 | 379.997 | 0.000 | -0.000 | 49.016 |
| -181.860 | 221.271 | -216.105 | -0.000 | 0.000 | 0.000 |
| -245.518 | 610.925 | -112.658 | -0.000 | 0.000 | 0.000 |
| -261.918 | 314.391 | 99.376 | 0.000 | 0.000 | 0.000 |
| -417.974 | 42.956 | -380.015 | 126.688 | 0.000 | 0.000 |
| -417.853 | 42.943 | -379.904 | 0.000 | 25.470 | 0.000 |
| -417.955 | 42.954 | -379.997 | -0.000 | 0.000 | 49.016 |
| 260.457 | 179.928 | 174.248 | 0.000 | 0.000 | -0.000 |
| 262.238 | 567.102 | 417.401 | 0.000 | -0.000 | -0.000 |
| 172.303 | 266.171 | 512.786 | 0.000 | 0.000 | 0.000 |
| 88.493 | 9.154 | 102.077 | 116.362 | 0.000 | 0.000 |
| 85.672 | 9.480 | 99.554 | 0.000 | 16.425 | 0.000 |
| 94.039 | 10.070 | 108.881 | 0.000 | 0.000 | 48.976 |
| 193.355 | 182.569 | 125.448 | -0.000 | 0.000 | 0.000 |
| 129.547 | 571.832 | 229.088 | -0.000 | 0.000 | 0.000 |
| 172.311 | 266.178 | 512.791 | -0.000 | -0.000 | -0.000 |
| -88.493 | -9.154 | -102.077 | 116.362 | -0.000 | -0.000 |
| -85.672 | -9.480 | -99.554 | -0.000 | 16.425 | -0.000 |
| -94.039 | -10.070 | -108.881 | -0.000 | -0.000 | 48.976 |
| 334.892 | 151.772 | 151.772 | -0.000 | 0.000 | -0.000 |
| 151.772 | 334.892 | 151.772 | -0.000 | 0.000 | 0.000 |
| 151.772 | 151.772 | 334.892 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 15.664 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 15.664 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 15.664 |
| 334.891 | 151.772 | 151.772 | 0.000 | -0.000 | -0.000 |
| 151.772 | 334.891 | 151.772 | -0.000 | 0.000 | 0.000 |
| 151.772 | 151.772 | 334.891 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 15.664 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 15.664 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 15.664 |
| 334.815 | 151.810 | 151.810 | 0.000 | -0.000 | 0.000 |
| 151.810 | 334.815 | 151.810 | -0.000 | 0.000 | 0.000 |
| 151.810 | 151.810 | 334.815 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 15.664 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 15.664 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 15.664 |
| 334.891 | 151.771 | 151.771 | -0.000 | 0.000 | -0.000 |
| 151.771 | 334.891 | 151.771 | -0.000 | 0.000 | -0.000 |
| 151.771 | 151.771 | 334.891 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 15.664 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 15.664 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 15.664 |
| 334.899 | 151.773 | 151.773 | -0.000 | 0.000 | 0.000 |
| 151.773 | 334.899 | 151.773 | -0.000 | 0.000 | -0.000 |
| 151.773 | 151.773 | 334.899 | -0.000 | 0.000 | 0.000 |
| 8471229.613 | 8467213.214 | 8069133.524 | -1217400.574 | -485801.717 | -313628.227 |
| 7916458.532 | 8008193.507 | 8126397.761 | -89235.823 | -1148112.567 | -430887.686 |
| 8241027.380 | 7398111.639 | 8246642.500 | -894370.418 | -327416.784 | -584513.517 |
| 334.807 | 151.809 | 151.809 | 0.000 | -0.000 | -0.000 |
| 151.809 | 334.807 | 151.809 | 0.000 | -0.000 | 0.000 |
| 151.809 | 151.809 | 334.807 | 0.000 | -0.000 | -0.000 |
| -7104594.242 | -7036187.314 | -7199817.353 | -688297.810 | -534370.387 | 167243.687 |
| -7170269.155 | -7507045.575 | -7499317.554 | 46937.097 | -597303.344 | 140933.112 |
| -8013666.015 | -7870568.141 | -7877947.673 | -163705.936 | 441622.803 | -1439385.693 |
| 56.192 | 66.992 | 66.992 | -0.000 | 0.000 | -0.000 |
| 66.992 | 56.192 | 66.992 | 0.000 | 0.000 | -0.000 |
| 66.992 | 66.992 | 56.192 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 63.596 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 63.596 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 63.596 |
| 56.956 | 66.610 | 66.610 | 0.000 | 0.000 | 0.000 |
| 66.610 | 56.956 | 66.610 | 0.000 | -0.000 | 0.000 |
| 66.610 | 66.610 | 56.956 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 63.596 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 63.596 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 63.596 |
| 56.956 | 66.610 | 66.610 | -0.000 | 0.000 | -0.000 |
| 66.610 | 56.956 | 66.610 | -0.000 | 0.000 | -0.000 |
| 66.610 | 66.610 | 56.956 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 63.596 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 63.596 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 63.596 |
| 56.956 | 66.610 | 66.610 | 0.000 | -0.000 | 0.000 |
| 66.610 | 56.956 | 66.610 | 0.000 | 0.000 | 0.000 |
| 66.610 | 66.610 | 56.956 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 63.596 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 63.596 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 63.596 |
| 56.954 | 66.610 | 66.610 | -0.000 | 0.000 | -0.000 |
| 66.610 | 56.954 | 66.610 | -0.000 | 0.000 | -0.000 |
| 66.610 | 66.610 | 56.954 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 63.596 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 63.596 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 63.596 |
| 56.959 | 66.610 | 66.610 | -0.000 | -0.000 | 0.000 |
| 66.610 | 56.959 | 66.610 | 0.000 | 0.000 | 0.000 |
| 66.610 | 66.610 | 56.959 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 63.596 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 63.596 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 63.596 |
| 303.974 | 342.062 | 342.062 | 0.000 | 0.000 | -0.000 |
| 342.062 | 303.974 | 342.062 | 0.000 | 0.000 | -0.000 |
| 342.062 | 342.062 | 303.974 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 175.457 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 175.457 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 175.457 |
| 303.974 | 342.062 | 342.062 | -0.000 | -0.000 | 0.000 |
| 342.062 | 303.974 | 342.062 | -0.000 | -0.000 | 0.000 |
| 342.062 | 342.062 | 303.974 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 175.457 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 175.457 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 175.457 |
| 303.973 | 342.061 | 342.061 | 0.000 | 0.000 | -0.000 |
| 342.061 | 303.973 | 342.061 | 0.000 | 0.000 | -0.000 |
| 342.061 | 342.061 | 303.973 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 175.457 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 175.457 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 175.457 |
| 303.974 | 342.062 | 342.062 | -0.000 | -0.000 | 0.000 |
| 342.062 | 303.974 | 342.062 | -0.000 | -0.000 | 0.000 |
| 342.062 | 342.062 | 303.974 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 175.457 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 175.457 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 175.457 |
| 303.969 | 342.055 | 342.055 | 0.000 | 0.000 | -0.000 |
| 342.055 | 303.969 | 342.055 | 0.000 | 0.000 | 0.000 |
| 342.055 | 342.055 | 303.969 | 0.000 | 0.000 | -0.000 |
| 0.001 | 0.001 | 0.001 | 117.132 | 0.000 | -0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | 117.132 | -0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | 0.000 | 117.132 |
| 303.978 | 342.068 | 342.068 | -0.000 | -0.000 | 0.000 |
| 342.068 | 303.978 | 342.068 | -0.000 | -0.000 | 0.000 |
| 342.068 | 342.068 | 303.978 | -0.000 | -0.000 | 0.000 |
| -0.001 | -0.001 | -0.001 | 117.132 | -0.000 | 0.000 |
| -0.001 | -0.001 | -0.001 | -0.000 | 117.132 | 0.000 |
| -0.001 | -0.001 | -0.001 | -0.000 | -0.000 | 117.132 |
| 114.814 | 33.773 | 33.773 | 0.000 | 0.000 | -0.000 |
| 33.773 | 114.814 | 33.773 | 0.000 | 0.000 | 0.000 |
| 33.773 | 33.773 | 114.814 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 33.063 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 33.063 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 33.063 |
| 114.814 | 33.773 | 33.773 | -0.000 | -0.000 | -0.000 |
| 33.773 | 114.814 | 33.773 | -0.000 | -0.000 | -0.000 |
| 33.773 | 33.773 | 114.814 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 33.063 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 33.063 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 33.063 |
| 114.814 | 33.773 | 33.773 | 0.000 | 0.000 | 0.000 |
| 33.773 | 114.814 | 33.773 | 0.000 | 0.000 | 0.000 |
| 33.773 | 33.773 | 114.814 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 33.162 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 33.162 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 33.162 |
| 114.814 | 33.773 | 33.773 | -0.000 | -0.000 | 0.000 |
| 33.773 | 114.814 | 33.773 | -0.000 | -0.000 | -0.000 |
| 33.773 | 33.773 | 114.814 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 33.162 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 33.162 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 33.162 |
| 114.813 | 33.773 | 33.773 | 0.000 | 0.000 | -0.000 |
| 33.773 | 114.813 | 33.773 | 0.000 | -0.000 | 0.000 |
| 33.773 | 33.773 | 114.813 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 33.063 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 33.063 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 33.063 |
| 114.814 | 33.774 | 33.774 | -0.000 | -0.000 | 0.000 |
| 33.774 | 114.814 | 33.774 | -0.000 | -0.000 | -0.000 |
| 33.774 | 33.774 | 114.814 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 33.063 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 33.063 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 33.063 |
| 330.692 | 152.303 | 152.303 | -0.000 | -0.000 | -0.000 |
| 152.303 | 330.692 | 152.303 | -0.000 | -0.000 | -0.000 |
| 152.303 | 152.303 | 330.692 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 51.766 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 51.766 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 51.766 |
| 330.692 | 152.303 | 152.303 | -0.000 | 0.000 | 0.000 |
| 152.303 | 330.692 | 152.303 | 0.000 | -0.000 | 0.000 |
| 152.303 | 152.303 | 330.692 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 51.766 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 51.766 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 51.766 |
| 330.692 | 152.303 | 152.303 | 0.000 | -0.000 | -0.000 |
| 152.303 | 330.692 | 152.303 | -0.000 | 0.000 | -0.000 |
| 152.303 | 152.303 | 330.692 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 51.766 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 51.766 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 51.766 |
| 330.692 | 152.303 | 152.303 | -0.000 | 0.000 | 0.000 |
| 152.303 | 330.692 | 152.303 | 0.000 | 0.000 | 0.000 |
| 152.303 | 152.303 | 330.692 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 51.766 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 51.766 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 51.766 |
| 330.691 | 152.304 | 152.304 | 0.000 | 0.000 | -0.000 |
| 152.304 | 330.691 | 152.304 | 0.000 | -0.000 | 0.000 |
| 152.304 | 152.304 | 330.691 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 51.766 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 51.766 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 51.766 |
| 330.693 | 152.302 | 152.302 | -0.000 | 0.000 | 0.000 |
| 152.302 | 330.693 | 152.302 | 0.000 | -0.000 | 0.000 |
| 152.302 | 152.302 | 330.693 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 51.766 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 51.766 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 51.766 |
| 61.453 | 20.970 | 20.970 | -0.000 | 0.000 | -0.000 |
| 20.970 | 61.453 | 20.970 | 0.000 | -0.000 | -0.000 |
| 20.970 | 20.970 | 61.453 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 20.981 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 20.981 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 20.981 |
| 61.453 | 20.970 | 20.970 | 0.000 | 0.000 | 0.000 |
| 20.970 | 61.453 | 20.970 | -0.000 | -0.000 | -0.000 |
| 20.970 | 20.970 | 61.453 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 20.981 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 20.981 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 20.981 |
| 61.453 | 20.970 | 20.970 | -0.000 | -0.000 | -0.000 |
| 20.970 | 61.453 | 20.970 | 0.000 | -0.000 | -0.000 |
| 20.970 | 20.970 | 61.453 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 20.981 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 20.981 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 20.981 |
| 61.453 | 20.970 | 20.970 | 0.000 | 0.000 | 0.000 |
| 20.970 | 61.453 | 20.970 | 0.000 | 0.000 | -0.000 |
| 20.970 | 20.970 | 61.453 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 20.981 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 20.981 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 20.981 |
| 61.452 | 20.970 | 20.970 | 0.000 | -0.000 | -0.000 |
| 20.970 | 61.452 | 20.970 | -0.000 | 0.000 | -0.000 |
| 20.970 | 20.970 | 61.452 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 20.981 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 20.981 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 20.981 |
| 61.453 | 20.970 | 20.970 | 0.000 | -0.000 | -0.000 |
| 20.970 | 61.453 | 20.970 | -0.000 | 0.000 | 0.000 |
| 20.970 | 20.970 | 61.453 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 20.981 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 20.981 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 20.981 |
| 293.358 | 224.550 | 170.002 | 0.000 | 0.000 | 0.000 |
| 224.550 | 293.358 | 170.002 | 0.000 | 0.000 | -0.000 |
| 170.002 | 170.002 | 346.232 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 68.571 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 68.571 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 69.052 |
| 348.913 | 168.995 | 170.002 | -0.000 | -0.000 | 0.000 |
| 168.995 | 348.913 | 170.002 | -0.000 | -0.000 | -0.000 |
| 170.002 | 170.002 | 346.232 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 68.571 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 68.571 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 69.052 |
| 348.913 | 168.995 | 170.002 | 0.000 | 0.000 | -0.000 |
| 168.995 | 348.913 | 170.002 | 0.000 | 0.000 | 0.000 |
| 170.002 | 170.002 | 346.232 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 68.571 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 68.571 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 69.052 |
| 348.913 | 168.995 | 170.002 | -0.000 | -0.000 | -0.000 |
| 168.995 | 348.913 | 170.002 | -0.000 | -0.000 | -0.000 |
| 170.002 | 170.002 | 346.233 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 68.571 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 68.571 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 69.052 |
| 348.911 | 168.994 | 170.001 | 0.000 | 0.000 | 0.000 |
| 168.994 | 348.911 | 170.001 | 0.000 | 0.000 | 0.000 |
| 170.001 | 170.001 | 346.229 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 56.988 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 56.988 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 69.052 |
| 348.916 | 168.995 | 170.002 | 0.000 | -0.000 | -0.000 |
| 168.995 | 348.916 | 170.002 | -0.000 | -0.000 | -0.000 |
| 170.003 | 170.003 | 346.235 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 56.988 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 56.988 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 69.052 |
| 192.791 | 291.197 | 304.682 | -0.000 | 0.000 | -0.000 |
| 239.733 | 672.439 | 233.602 | -0.000 | 0.000 | 0.000 |
| 24.623 | 344.277 | 576.929 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 89.892 | 0.000 | 0.000 |
| -130.820 | 56.577 | 123.074 | -0.000 | 52.434 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 97.612 |
| 395.524 | 237.547 | 224.282 | 0.000 | -0.000 | -0.000 |
| 368.195 | 610.464 | 103.859 | 0.000 | -0.000 | 0.000 |
| 225.295 | 232.430 | 408.795 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 89.892 | -0.000 | 0.000 |
| 130.820 | -56.577 | -123.074 | 0.000 | 52.434 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 97.612 |
| 300.695 | 240.824 | 191.148 | -0.000 | 0.000 | -0.000 |
| 213.478 | 679.878 | 244.939 | -0.000 | -0.000 | -0.000 |
| 225.295 | 232.430 | 408.796 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 89.892 | -0.000 | -0.000 |
| -13.539 | 5.855 | 12.737 | -0.000 | 51.231 | -0.000 |
| -13.107 | 5.668 | 12.331 | -0.000 | 0.000 | 93.928 |
| 395.523 | 237.547 | 224.282 | 0.000 | 0.000 | -0.000 |
| 250.347 | 661.311 | 214.564 | 0.000 | 0.000 | 0.000 |
| 46.275 | 178.239 | 124.550 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 89.892 | 0.000 | 0.000 |
| 13.539 | -5.855 | -12.737 | 0.000 | 51.231 | 0.000 |
| 13.107 | -5.668 | -12.331 | 0.000 | -0.000 | 93.928 |
| 232.940 | 232.890 | 134.151 | -0.000 | 0.000 | -0.000 |
| 239.734 | 672.437 | 233.603 | -0.000 | -0.000 | 0.000 |
| 110.157 | 217.210 | 253.850 | -0.000 | 0.000 | -0.000 |
| -1.251 | 0.542 | 1.178 | 87.999 | 0.000 | -0.000 |
| -0.766 | 1.130 | 2.680 | 0.000 | 33.255 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 97.612 |
| -93.986 | 170.232 | -169.240 | 0.000 | 0.000 | 0.000 |
| 238.502 | 667.275 | 226.869 | 0.000 | -0.000 | 0.000 |
| -298.387 | 147.596 | -179.706 | 0.000 | 0.000 | 0.000 |
| 1.251 | -0.542 | -1.178 | 87.999 | -0.000 | -0.000 |
| 0.766 | -1.130 | -2.680 | 0.000 | 33.255 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 97.612 |
| 479.660 | 224.561 | 1061.350 | -0.000 | 0.000 | -0.000 |
| 226.349 | 542.847 | 219.089 | -0.000 | -0.000 | -0.000 |
| 174.786 | 293.454 | 828.797 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 94.816 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 122.160 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 103.337 |
| 438.572 | 226.349 | 244.058 | 0.000 | -0.000 | -0.000 |
| 226.348 | 542.847 | 219.089 | 0.000 | -0.000 | -0.000 |
| 244.059 | 219.090 | 425.670 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 94.816 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 122.160 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 103.337 |
| 402.249 | 231.533 | 269.063 | -0.000 | -0.000 | -0.000 |
| 225.994 | 538.631 | 295.605 | -0.000 | 0.000 | 0.000 |
| 215.583 | 210.338 | 463.181 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 94.816 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 122.160 | 0.000 |
| -4.152 | 5.767 | 38.288 | -0.000 | 0.000 | 100.281 |
| 438.572 | 226.349 | 244.058 | 0.000 | 0.000 | 0.000 |
| 226.348 | 542.847 | 219.089 | 0.000 | -0.000 | -0.000 |
| 234.850 | 209.962 | 362.912 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 94.816 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 122.160 | -0.000 |
| 4.152 | -5.767 | -38.288 | 0.000 | -0.000 | 100.281 |
| 400.909 | 226.374 | 233.352 | -0.000 | 0.000 | -0.000 |
| 226.349 | 542.845 | 219.090 | -0.000 | -0.000 | -0.000 |
| 244.059 | 219.090 | 425.671 | -0.000 | 0.000 | 0.000 |
| 1.019 | 0.075 | 8.463 | 92.442 | -0.000 | 0.000 |
| -0.453 | 0.628 | 4.171 | -0.000 | 107.176 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 103.337 |
| 375.347 | 216.146 | 197.828 | 0.000 | 0.000 | 0.000 |
| 226.223 | 536.790 | 212.854 | 0.000 | 0.000 | 0.000 |
| 225.980 | 213.798 | 387.546 | 0.000 | -0.000 | 0.000 |
| -1.019 | -0.075 | -8.463 | 92.442 | -0.000 | -0.000 |
| 0.453 | -0.628 | -4.171 | 0.000 | 107.176 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 103.337 |
| 870.896 | 95.396 | 618.051 | -0.000 | 0.000 | 0.000 |
| 239.764 | 666.854 | 234.121 | -0.000 | 0.000 | -0.000 |
| 717.038 | 87.926 | 794.421 | -0.000 | 0.000 | -0.000 |
| 529.852 | -142.882 | 430.449 | 89.709 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 87.504 | -0.000 |
| 5704.574 | 664.234 | 5168.311 | -0.000 | -0.000 | 93.847 |
| -191.673 | 381.362 | -244.740 | 0.000 | -0.000 | -0.000 |
| -1342.555 | 654.319 | -1144.468 | 0.000 | -0.000 | -0.000 |
| 224.825 | 232.596 | 411.948 | 0.000 | 0.000 | 0.000 |
| -529.852 | 142.882 | -430.449 | 89.709 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 87.504 | -0.000 |
| -4062.829 | -402.835 | -3487.482 | 0.000 | -0.000 | 92.873 |
| 399.722 | 237.350 | 223.937 | -0.000 | -0.000 | -0.000 |
| 239.764 | 666.854 | 234.121 | -0.000 | -0.000 | -0.000 |
| 247.010 | 215.318 | 413.507 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 91.106 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 87.504 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 99.010 |
| 399.721 | 237.350 | 223.936 | 0.000 | -0.000 | -0.000 |
| 180.172 | 678.370 | 182.509 | 0.000 | 0.000 | -0.000 |
| -886.318 | 73.166 | -698.859 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 91.106 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 87.504 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 99.010 |
| 399.723 | 237.351 | 223.937 | -0.000 | -0.000 | -0.000 |
| 239.765 | 666.853 | 234.122 | -0.000 | -0.000 | 0.000 |
| 224.826 | 232.597 | 411.949 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 91.106 | -0.000 | -0.000 |
| 11.642 | 0.294 | 10.156 | -0.000 | 35.004 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 99.010 |
| -74.513 | 174.473 | -150.752 | 0.000 | -0.000 | -0.000 |
| 105.337 | 644.644 | 105.258 | 0.000 | -0.000 | -0.000 |
| 118.774 | 223.795 | 271.284 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 91.106 | -0.000 | 0.000 |
| -11.642 | -0.294 | -10.156 | 0.000 | 35.004 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 99.010 |
| 740.379 | 820.062 | 820.062 | -0.000 | 0.000 | 0.000 |
| 820.062 | 740.379 | 820.062 | 0.000 | 0.000 | -0.000 |
| 820.062 | 820.062 | 740.379 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 145.409 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 145.409 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 145.409 |
| 740.381 | 820.064 | 820.064 | 0.000 | -0.000 | 0.000 |
| 820.064 | 740.381 | 820.064 | -0.000 | 0.000 | 0.000 |
| 820.064 | 820.064 | 740.381 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 145.409 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 145.409 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 145.409 |
| 740.373 | 820.056 | 820.056 | -0.000 | -0.000 | -0.000 |
| 820.056 | 740.373 | 820.056 | -0.000 | -0.000 | 0.000 |
| 820.056 | 820.056 | 740.373 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 145.409 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 145.409 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 145.409 |
| 740.387 | 820.070 | 820.070 | 0.000 | 0.000 | -0.000 |
| 820.070 | 740.387 | 820.070 | 0.000 | 0.000 | 0.000 |
| 820.070 | 820.070 | 740.387 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 145.409 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 145.409 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 145.409 |
| 740.312 | 819.994 | 819.994 | -0.000 | -0.000 | -0.000 |
| 819.994 | 740.312 | 819.994 | -0.000 | -0.000 | -0.000 |
| 819.994 | 819.994 | 740.312 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 145.409 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 145.409 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 145.409 |
| 740.448 | 820.132 | 820.132 | 0.000 | 0.000 | -0.000 |
| 820.132 | 740.448 | 820.132 | 0.000 | 0.000 | 0.000 |
| 820.132 | 820.132 | 740.448 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 145.409 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 145.409 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 145.409 |
| -150.486 | -145.660 | -145.660 | 0.000 | 0.000 | -0.000 |
| -145.660 | -150.486 | -145.660 | -0.000 | 0.000 | 0.000 |
| -145.660 | -145.660 | -150.486 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -145.660 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -145.660 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | -145.660 |
| -150.486 | -145.660 | -145.660 | -0.000 | -0.000 | 0.000 |
| -145.660 | -150.486 | -145.660 | -0.000 | -0.000 | -0.000 |
| -145.660 | -145.660 | -150.486 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -145.660 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -145.660 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -145.660 |
| -150.486 | -145.660 | -145.660 | -0.000 | -0.000 | 0.000 |
| -145.660 | -150.486 | -145.660 | 0.000 | 0.000 | 0.000 |
| -145.660 | -145.660 | -150.486 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -145.660 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -145.660 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -145.660 |
| -150.486 | -145.660 | -145.660 | 0.000 | -0.000 | -0.000 |
| -145.660 | -150.486 | -145.660 | -0.000 | -0.000 | 0.000 |
| -145.660 | -145.660 | -150.486 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -145.660 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -145.660 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -145.660 |
| -150.485 | -145.660 | -145.660 | -0.000 | 0.000 | -0.000 |
| -145.660 | -150.485 | -145.660 | 0.000 | -0.000 | -0.000 |
| -145.660 | -145.660 | -150.485 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -145.660 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -145.660 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -145.660 |
| -150.486 | -145.659 | -145.659 | -0.000 | 0.000 | 0.000 |
| -145.659 | -150.486 | -145.659 | -0.000 | -0.000 | 0.000 |
| -145.659 | -145.659 | -150.486 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -145.660 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -145.660 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -145.660 |
| -96.703 | -72.820 | -72.820 | -0.000 | 0.000 | 0.000 |
| -72.820 | -96.703 | -72.820 | 0.000 | 0.000 | 0.000 |
| -72.820 | -72.820 | -96.703 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.035 | -0.035 | -92.694 | 0.000 | 0.000 |
| -0.035 | -0.000 | -0.035 | -0.000 | -92.694 | 0.000 |
| -0.035 | -0.035 | -0.000 | 0.000 | 0.000 | -92.694 |
| -96.563 | -72.750 | -72.750 | 0.000 | -0.000 | -0.000 |
| -72.750 | -96.563 | -72.750 | 0.000 | -0.000 | 0.000 |
| -72.750 | -72.750 | -96.563 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.035 | 0.035 | -92.694 | -0.000 | -0.000 |
| 0.035 | 0.000 | 0.035 | 0.000 | -92.694 | -0.000 |
| 0.035 | 0.035 | 0.000 | 0.000 | -0.000 | -92.694 |
| -96.703 | -72.820 | -72.820 | -0.000 | 0.000 | 0.000 |
| -72.820 | -96.703 | -72.820 | -0.000 | 0.000 | 0.000 |
| -72.820 | -72.820 | -96.703 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.035 | -0.035 | -92.694 | 0.000 | 0.000 |
| -0.035 | -0.000 | -0.035 | -0.000 | -92.694 | 0.000 |
| -0.035 | -0.035 | -0.000 | -0.000 | 0.000 | -92.694 |
| -96.563 | -72.750 | -72.750 | -0.000 | -0.000 | -0.000 |
| -72.750 | -96.563 | -72.750 | -0.000 | -0.000 | -0.000 |
| -72.750 | -72.750 | -96.563 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.035 | 0.035 | -92.694 | -0.000 | 0.000 |
| 0.035 | 0.000 | 0.035 | -0.000 | -92.694 | -0.000 |
| 0.035 | 0.035 | 0.000 | 0.000 | -0.000 | -92.694 |
| -96.702 | -72.820 | -72.820 | -0.000 | 0.000 | 0.000 |
| -72.820 | -96.702 | -72.820 | -0.000 | 0.000 | 0.000 |
| -72.820 | -72.820 | -96.702 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.035 | -0.035 | -92.694 | 0.000 | 0.000 |
| -0.035 | -0.000 | -0.035 | -0.000 | -92.694 | 0.000 |
| -0.035 | -0.035 | -0.000 | -0.000 | 0.000 | -92.694 |
| -96.564 | -72.749 | -72.749 | -0.000 | -0.000 | -0.000 |
| -72.749 | -96.564 | -72.749 | 0.000 | -0.000 | -0.000 |
| -72.749 | -72.749 | -96.564 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.035 | 0.035 | -92.694 | 0.000 | -0.000 |
| 0.035 | 0.000 | 0.035 | -0.000 | -92.694 | -0.000 |
| 0.035 | 0.035 | 0.000 | -0.000 | -0.000 | -92.694 |
| 72.680 | 36.422 | 36.422 | -0.000 | -0.000 | -0.000 |
| 36.422 | 72.680 | 36.422 | -0.000 | -0.000 | -0.000 |
| 36.422 | 36.422 | 72.680 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 36.340 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 36.340 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 36.340 |
| 72.680 | 36.422 | 36.422 | 0.000 | 0.000 | 0.000 |
| 36.422 | 72.680 | 36.422 | 0.000 | 0.000 | 0.000 |
| 36.422 | 36.422 | 72.680 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 36.340 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 36.340 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 36.340 |
| 72.680 | 36.422 | 36.422 | -0.000 | -0.000 | -0.000 |
| 36.422 | 72.680 | 36.422 | -0.000 | -0.000 | -0.000 |
| 36.422 | 36.422 | 72.680 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 36.340 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 36.340 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 36.340 |
| 72.680 | 36.422 | 36.422 | 0.000 | 0.000 | 0.000 |
| 36.422 | 72.680 | 36.422 | 0.000 | 0.000 | 0.000 |
| 36.422 | 36.422 | 72.680 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 36.340 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 36.340 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 36.340 |
| 72.680 | 36.422 | 36.422 | -0.000 | -0.000 | -0.000 |
| 36.422 | 72.680 | 36.422 | -0.000 | -0.000 | -0.000 |
| 36.422 | 36.422 | 72.680 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 36.340 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 36.340 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 36.340 |
| 72.680 | 36.422 | 36.422 | 0.000 | 0.000 | 0.000 |
| 36.422 | 72.680 | 36.422 | 0.000 | 0.000 | 0.000 |
| 36.422 | 36.422 | 72.680 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 36.340 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 36.340 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 36.340 |
| 888.542 | 645.233 | 645.233 | -0.000 | -0.000 | -0.000 |
| 645.233 | 888.542 | 645.233 | -0.000 | -0.000 | 0.000 |
| 645.233 | 645.233 | 888.542 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 9.223 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 9.223 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 9.223 |
| 888.545 | 645.231 | 645.231 | -0.000 | 0.000 | 0.000 |
| 645.231 | 888.545 | 645.231 | 0.000 | 0.000 | 0.000 |
| 645.231 | 645.231 | 888.545 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 9.223 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 9.223 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 9.223 |
| 888.541 | 645.232 | 645.232 | -0.000 | -0.000 | 0.000 |
| 645.232 | 888.541 | 645.232 | -0.000 | -0.000 | 0.000 |
| 645.232 | 645.232 | 888.541 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 9.223 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 9.223 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 9.223 |
| 888.546 | 645.232 | 645.232 | 0.000 | 0.000 | -0.000 |
| 645.232 | 888.546 | 645.232 | -0.000 | -0.000 | -0.000 |
| 645.232 | 645.232 | 888.546 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 9.223 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 9.223 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 9.223 |
| 888.537 | 645.225 | 645.225 | -0.000 | -0.000 | 0.000 |
| 645.225 | 888.537 | 645.225 | -0.000 | 0.000 | 0.000 |
| 645.225 | 645.225 | 888.537 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.001 | -0.001 | 9.086 | -0.000 | 0.000 |
| -0.001 | -0.000 | -0.001 | -0.000 | 9.086 | 0.000 |
| -0.001 | -0.001 | -0.000 | -0.000 | -0.000 | 9.086 |
| 888.550 | 645.239 | 645.239 | -0.000 | 0.000 | -0.000 |
| 645.239 | 888.550 | 645.239 | 0.000 | 0.000 | 0.000 |
| 645.239 | 645.239 | 888.550 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.001 | 0.001 | 9.086 | 0.000 | -0.000 |
| 0.001 | 0.000 | 0.001 | 0.000 | 9.086 | -0.000 |
| 0.001 | 0.001 | 0.000 | 0.000 | 0.000 | 9.086 |
| -370.187 | -112.479 | -112.479 | 0.000 | -0.000 | 0.000 |
| -112.479 | -370.187 | -112.479 | -0.000 | -0.000 | 0.000 |
| -112.479 | -112.479 | -370.187 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -215.810 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -215.810 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -215.810 |
| -370.187 | -112.478 | -112.478 | 0.000 | 0.000 | -0.000 |
| -112.478 | -370.187 | -112.478 | -0.000 | 0.000 | -0.000 |
| -112.478 | -112.478 | -370.187 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -215.810 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -215.810 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | -215.810 |
| -370.187 | -112.479 | -112.479 | -0.000 | -0.000 | 0.000 |
| -112.479 | -370.187 | -112.479 | -0.000 | 0.000 | 0.000 |
| -112.479 | -112.479 | -370.187 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -112.478 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -112.478 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -112.478 |
| -370.187 | -112.478 | -112.478 | 0.000 | 0.000 | -0.000 |
| -112.478 | -370.187 | -112.478 | 0.000 | 0.000 | -0.000 |
| -112.478 | -112.478 | -370.187 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -112.478 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -112.478 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -112.478 |
| -370.188 | -112.479 | -112.479 | -0.000 | -0.000 | 0.000 |
| -112.479 | -370.188 | -112.479 | -0.000 | -0.000 | 0.000 |
| -112.479 | -112.479 | -370.188 | 0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -112.478 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -112.478 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -112.478 |
| -370.186 | -112.478 | -112.478 | 0.000 | -0.000 | -0.000 |
| -112.478 | -370.186 | -112.478 | 0.000 | 0.000 | -0.000 |
| -112.478 | -112.478 | -370.186 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -112.478 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -112.478 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -112.478 |
| -246.951 | -56.537 | -56.537 | 0.000 | -0.000 | 0.000 |
| -56.537 | -246.951 | -56.537 | 0.000 | -0.000 | 0.000 |
| -56.537 | -56.537 | -246.951 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -151.454 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -151.454 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -151.454 |
| -246.951 | -56.537 | -56.537 | -0.000 | 0.000 | -0.000 |
| -56.537 | -246.951 | -56.537 | -0.000 | 0.000 | -0.000 |
| -56.537 | -56.537 | -246.951 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -151.454 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -151.454 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -151.454 |
| -310.514 | -24.756 | -24.756 | 0.000 | -0.000 | 0.000 |
| -24.756 | -310.514 | -24.756 | 0.000 | -0.000 | 0.000 |
| -24.756 | -24.756 | -310.514 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -87.891 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -87.891 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | -87.891 |
| -246.951 | -56.537 | -56.537 | -0.000 | 0.000 | -0.000 |
| -56.537 | -246.951 | -56.537 | -0.000 | 0.000 | -0.000 |
| -56.537 | -56.537 | -246.951 | -0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -87.891 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -87.891 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | -87.891 |
| -246.952 | -56.538 | -56.538 | 0.000 | -0.000 | -0.000 |
| -56.538 | -246.952 | -56.538 | 0.000 | -0.000 | 0.000 |
| -56.538 | -56.538 | -246.952 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -87.891 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -87.891 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -87.891 |
| -246.949 | -56.537 | -56.537 | -0.000 | 0.000 | -0.000 |
| -56.537 | -246.949 | -56.537 | -0.000 | 0.000 | -0.000 |
| -56.537 | -56.537 | -246.949 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -87.891 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -87.891 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -87.891 |
| 86.293 | 24.332 | 24.332 | -0.000 | -0.000 | -0.000 |
| 24.332 | 86.293 | 24.332 | -0.000 | -0.000 | -0.000 |
| 24.332 | 24.332 | 86.293 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 22.466 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 22.466 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 22.466 |
| 87.331 | 23.813 | 23.813 | 0.000 | -0.000 | 0.000 |
| 23.813 | 87.331 | 23.813 | -0.000 | 0.000 | 0.000 |
| 23.813 | 23.813 | 87.331 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 22.466 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 22.466 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 22.466 |
| 87.331 | 23.813 | 23.813 | -0.000 | 0.000 | -0.000 |
| 23.813 | 87.331 | 23.813 | -0.000 | -0.000 | -0.000 |
| 23.813 | 23.813 | 87.331 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 22.466 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 22.466 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 22.466 |
| 86.293 | 24.332 | 24.332 | -0.000 | -0.000 | 0.000 |
| 24.332 | 86.293 | 24.332 | 0.000 | 0.000 | -0.000 |
| 24.332 | 24.332 | 86.293 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 22.466 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 22.466 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 22.466 |
| 87.331 | 23.813 | 23.813 | -0.000 | -0.000 | -0.000 |
| 23.813 | 87.331 | 23.813 | -0.000 | -0.000 | 0.000 |
| 23.813 | 23.813 | 87.331 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 22.466 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 22.466 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 22.466 |
| 87.331 | 23.813 | 23.813 | 0.000 | 0.000 | 0.000 |
| 23.813 | 87.331 | 23.813 | 0.000 | 0.000 | 0.000 |
| 23.813 | 23.813 | 87.331 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 22.466 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 22.466 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 22.466 |
| 558.691 | 518.522 | 518.522 | -0.000 | -0.000 | 0.000 |
| 518.522 | 558.691 | 518.522 | -0.000 | -0.000 | 0.000 |
| 518.522 | 518.522 | 558.691 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 114.424 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 114.424 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 114.424 |
| 558.691 | 518.523 | 518.523 | -0.000 | 0.000 | -0.000 |
| 518.523 | 558.691 | 518.523 | -0.000 | 0.000 | 0.000 |
| 518.523 | 518.523 | 558.691 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 114.424 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 114.424 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 114.424 |
| 558.689 | 518.521 | 518.521 | -0.000 | -0.000 | 0.000 |
| 518.521 | 558.689 | 518.521 | -0.000 | -0.000 | 0.000 |
| 518.521 | 518.521 | 558.689 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 114.424 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 114.424 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 114.424 |
| 558.693 | 518.525 | 518.525 | -0.000 | 0.000 | -0.000 |
| 518.525 | 558.693 | 518.525 | 0.000 | 0.000 | 0.000 |
| 518.525 | 518.525 | 558.693 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 114.424 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 114.424 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 114.424 |
| 558.672 | 518.503 | 518.503 | 0.000 | -0.000 | 0.000 |
| 518.503 | 558.672 | 518.503 | 0.000 | -0.000 | 0.000 |
| 518.503 | 518.503 | 558.672 | -0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 114.424 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 114.424 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 114.424 |
| 558.710 | 518.542 | 518.542 | 0.000 | 0.000 | -0.000 |
| 518.542 | 558.710 | 518.542 | -0.000 | 0.000 | -0.000 |
| 518.542 | 518.542 | 558.710 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 114.424 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 114.424 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 114.424 |
| -192.102 | -132.870 | -132.870 | 0.000 | 0.000 | -0.000 |
| -132.870 | -192.102 | -132.870 | 0.000 | 0.000 | -0.000 |
| -132.870 | -132.870 | -192.102 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -132.870 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -132.870 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -132.870 |
| -192.102 | -132.870 | -132.870 | -0.000 | -0.000 | -0.000 |
| -132.870 | -192.102 | -132.870 | 0.000 | -0.000 | 0.000 |
| -132.870 | -132.870 | -192.102 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -132.870 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -132.870 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -132.870 |
| -192.102 | -132.870 | -132.870 | 0.000 | 0.000 | -0.000 |
| -132.870 | -192.102 | -132.870 | 0.000 | 0.000 | -0.000 |
| -132.870 | -132.870 | -192.102 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -132.870 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -132.870 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | -132.870 |
| -192.102 | -132.870 | -132.870 | 0.000 | 0.000 | 0.000 |
| -132.870 | -192.102 | -132.870 | 0.000 | -0.000 | 0.000 |
| -132.870 | -132.870 | -192.102 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -132.870 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -132.870 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -132.870 |
| -192.103 | -132.870 | -132.870 | 0.000 | 0.000 | -0.000 |
| -132.870 | -192.103 | -132.870 | 0.000 | 0.000 | -0.000 |
| -132.870 | -132.870 | -192.103 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -132.870 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -132.870 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | -132.870 |
| -192.101 | -132.870 | -132.870 | -0.000 | -0.000 | 0.000 |
| -132.870 | -192.101 | -132.870 | -0.000 | -0.000 | 0.000 |
| -132.870 | -132.870 | -192.101 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -132.870 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -132.870 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -132.870 |
| -143.358 | -105.611 | -105.611 | -0.000 | 0.000 | -0.000 |
| -105.611 | -143.358 | -105.611 | -0.000 | -0.000 | -0.000 |
| -105.611 | -105.611 | -143.358 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -128.241 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -128.241 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -128.241 |
| -143.358 | -105.611 | -105.611 | 0.000 | 0.000 | 0.000 |
| -105.611 | -143.358 | -105.611 | 0.000 | 0.000 | 0.000 |
| -105.611 | -105.611 | -143.358 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -128.241 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -128.241 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -128.241 |
| -143.358 | -105.611 | -105.611 | 0.000 | -0.000 | -0.000 |
| -105.611 | -143.358 | -105.611 | -0.000 | 0.000 | -0.000 |
| -105.611 | -105.611 | -143.358 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -128.241 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -128.241 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -128.241 |
| -143.358 | -105.611 | -105.611 | 0.000 | 0.000 | 0.000 |
| -105.611 | -143.358 | -105.611 | 0.000 | -0.000 | 0.000 |
| -105.611 | -105.611 | -143.358 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -128.241 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -128.241 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -128.241 |
| -143.359 | -105.611 | -105.611 | -0.000 | 0.000 | -0.000 |
| -105.611 | -143.359 | -105.611 | -0.000 | 0.000 | -0.000 |
| -105.611 | -105.611 | -143.359 | -0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -128.241 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -128.241 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -128.241 |
| -143.358 | -105.611 | -105.611 | 0.000 | 0.000 | 0.000 |
| -105.611 | -143.358 | -105.611 | 0.000 | -0.000 | 0.000 |
| -105.611 | -105.611 | -143.358 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -128.241 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -128.241 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -128.241 |
| 47.425 | 34.732 | 34.732 | -0.000 | -0.000 | -0.000 |
| 34.732 | 47.425 | 34.732 | -0.000 | -0.000 | 0.000 |
| 34.732 | 34.732 | 47.425 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 34.647 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 34.647 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 34.647 |
| 47.425 | 34.732 | 34.732 | 0.000 | 0.000 | 0.000 |
| 34.732 | 47.425 | 34.732 | 0.000 | 0.000 | 0.000 |
| 34.732 | 34.732 | 47.425 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 34.647 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 34.647 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 34.647 |
| 47.425 | 34.732 | 34.732 | -0.000 | -0.000 | -0.000 |
| 34.732 | 47.425 | 34.732 | -0.000 | -0.000 | -0.000 |
| 34.732 | 34.732 | 47.425 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 34.647 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 34.647 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 34.647 |
| 47.425 | 34.732 | 34.732 | 0.000 | 0.000 | 0.000 |
| 34.732 | 47.425 | 34.732 | 0.000 | 0.000 | 0.000 |
| 34.732 | 34.732 | 47.425 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 34.647 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 34.647 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 34.647 |
| 47.425 | 34.732 | 34.732 | -0.000 | -0.000 | -0.000 |
| 34.732 | 47.425 | 34.732 | -0.000 | -0.000 | -0.000 |
| 34.732 | 34.732 | 47.425 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 34.647 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 34.647 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 34.647 |
| 47.425 | 34.732 | 34.732 | 0.000 | 0.000 | 0.000 |
| 34.732 | 47.425 | 34.732 | 0.000 | 0.000 | 0.000 |
| 34.732 | 34.732 | 47.425 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 34.647 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 34.647 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 34.647 |
| 830.801 | 685.696 | 612.952 | 0.000 | 0.000 | 0.000 |
| 685.696 | 830.801 | 612.952 | 0.000 | -0.000 | 0.000 |
| 612.952 | 612.952 | 939.326 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 10.085 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 10.085 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 72.553 |
| 512.812 | 1003.684 | 612.951 | 0.000 | 0.000 | -0.000 |
| 685.695 | 830.801 | 612.951 | 0.000 | 0.000 | -0.000 |
| 612.951 | 612.951 | 939.325 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 10.085 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 10.085 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 72.553 |
| 830.802 | 685.696 | 612.952 | -0.000 | -0.000 | 0.000 |
| 685.696 | 830.802 | 612.952 | 0.000 | 0.000 | 0.000 |
| 612.953 | 612.953 | 939.327 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 10.085 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 10.085 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 72.553 |
| 563.764 | 952.731 | 612.950 | 0.000 | -0.000 | -0.000 |
| 858.811 | 657.683 | 612.950 | -0.000 | -0.000 | -0.000 |
| 612.950 | 612.950 | 939.324 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 10.085 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 10.085 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 72.553 |
| 538.305 | 978.214 | 612.967 | 0.000 | -0.000 | 0.000 |
| 882.297 | 634.218 | 612.963 | 0.000 | -0.000 | -0.000 |
| 612.963 | 612.963 | 939.336 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 10.085 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 10.085 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 72.553 |
| 610.713 | 905.765 | 612.939 | -0.000 | 0.000 | -0.000 |
| 772.248 | 744.234 | 612.942 | 0.000 | 0.000 | -0.000 |
| 612.940 | 612.940 | 939.314 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 10.085 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 10.085 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 72.553 |
| -295.297 | -98.432 | -43.866 | 0.000 | -0.000 | 0.000 |
| -98.432 | -295.297 | -43.866 | 0.000 | 0.000 | 0.000 |
| -43.866 | -43.866 | -308.589 | -0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -43.866 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -43.866 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -98.432 |
| -295.297 | -98.432 | -43.866 | -0.000 | -0.000 | 0.000 |
| -98.432 | -295.297 | -43.866 | -0.000 | -0.000 | -0.000 |
| -43.866 | -43.866 | -308.589 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -43.866 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -43.866 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -98.432 |
| -295.297 | -98.432 | -43.866 | 0.000 | 0.000 | -0.000 |
| -98.432 | -295.297 | -43.866 | 0.000 | 0.000 | -0.000 |
| -43.866 | -43.866 | -308.589 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -43.866 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -43.866 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -314.713 |
| -511.577 | 117.849 | -43.866 | -0.000 | -0.000 | -0.000 |
| -98.432 | -295.296 | -43.866 | -0.000 | 0.000 | -0.000 |
| -43.866 | -43.866 | -308.589 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -43.866 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -43.866 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -314.713 |
| -295.298 | -98.433 | -43.866 | 0.000 | -0.000 | 0.000 |
| -87.808 | -305.923 | -43.866 | 0.000 | 0.000 | 0.000 |
| -43.866 | -43.866 | -308.590 | -0.000 | -0.000 | 0.000 |
| 0.001 | -0.001 | -0.000 | -43.866 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -43.866 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -101.812 |
| -302.054 | -91.673 | -43.866 | -0.000 | -0.000 | -0.000 |
| -80.579 | -313.148 | -43.866 | -0.000 | -0.000 | -0.000 |
| -43.866 | -43.866 | -308.588 | 0.000 | -0.000 | -0.000 |
| -0.001 | 0.001 | 0.000 | -43.866 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -43.866 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -101.812 |
| -287.684 | 38.416 | -21.322 | 0.000 | -0.000 | -0.000 |
| 38.416 | -287.684 | -21.322 | 0.000 | -0.000 | 0.000 |
| -20.017 | -20.017 | -228.889 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -35.961 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -35.961 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -0.000 | -163.050 |
| -1036.351 | 787.083 | -21.322 | -0.000 | 0.000 | 0.000 |
| 38.416 | -287.684 | -21.322 | -0.000 | 0.000 | 0.000 |
| -20.017 | -20.017 | -228.889 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -35.961 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -35.961 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | -163.050 |
| -287.685 | 38.416 | -21.322 | 0.000 | -0.000 | -0.000 |
| -53.692 | -195.576 | -21.322 | 0.000 | -0.000 | -0.000 |
| -20.017 | -20.017 | -228.889 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -35.961 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -35.961 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -70.942 |
| -662.017 | 412.750 | -21.321 | -0.000 | 0.000 | 0.000 |
| -53.692 | -195.576 | -21.322 | -0.000 | 0.000 | 0.000 |
| -20.017 | -20.017 | -228.889 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -35.961 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -35.961 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -70.942 |
| -204.917 | -44.353 | -21.322 | 0.000 | -0.000 | -0.000 |
| -53.693 | -195.577 | -21.322 | 0.000 | -0.000 | -0.000 |
| -20.017 | -20.017 | -228.890 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -35.961 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -35.961 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -70.942 |
| -207.088 | -42.179 | -21.322 | -0.000 | 0.000 | -0.000 |
| -53.692 | -195.575 | -21.322 | -0.000 | 0.000 | 0.000 |
| -20.016 | -20.016 | -228.888 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -35.961 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -35.961 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -70.942 |
| 90.891 | 30.352 | 24.281 | -0.000 | 0.000 | 0.000 |
| 33.397 | 87.846 | 24.281 | -0.000 | 0.000 | -0.000 |
| 24.281 | 24.281 | 96.962 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 24.199 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 24.199 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 27.224 |
| 87.846 | 33.397 | 24.281 | 0.000 | -0.000 | 0.000 |
| 33.397 | 87.846 | 24.281 | 0.000 | -0.000 | 0.000 |
| 24.281 | 24.281 | 96.962 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 24.199 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 24.199 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 27.224 |
| 90.891 | 30.352 | 24.281 | 0.000 | 0.000 | -0.000 |
| 30.352 | 90.891 | 24.281 | 0.000 | 0.000 | 0.000 |
| 24.281 | 24.281 | 96.962 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 24.199 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 24.199 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 30.270 |
| 90.891 | 30.352 | 24.281 | -0.000 | -0.000 | -0.000 |
| 30.351 | 90.891 | 24.281 | 0.000 | -0.000 | -0.000 |
| 24.281 | 24.281 | 96.962 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 24.199 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 24.199 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 30.270 |
| 90.891 | 30.352 | 24.281 | 0.000 | 0.000 | -0.000 |
| 33.397 | 87.846 | 24.281 | -0.000 | 0.000 | -0.000 |
| 24.281 | 24.281 | 96.962 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 24.199 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 24.199 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 30.270 |
| 90.891 | 30.351 | 24.281 | 0.000 | -0.000 | -0.000 |
| 30.351 | 90.891 | 24.281 | 0.000 | -0.000 | -0.000 |
| 24.281 | 24.281 | 96.962 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 24.199 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 24.199 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 30.270 |
| 770.383 | 641.862 | 560.131 | -0.000 | 0.000 | -0.000 |
| 867.762 | 544.483 | 560.131 | -0.000 | -0.000 | 0.000 |
| 560.131 | 560.131 | 935.494 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 5.927 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 5.927 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -256.925 |
| 770.384 | 641.862 | 560.131 | -0.000 | -0.000 | 0.000 |
| 641.862 | 770.384 | 560.131 | 0.000 | 0.000 | -0.000 |
| 560.131 | 560.131 | 935.495 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 5.927 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 5.927 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -256.925 |
| 770.380 | 641.859 | 560.128 | -0.000 | -0.000 | 0.000 |
| 641.859 | 770.380 | 560.128 | 0.000 | -0.000 | -0.000 |
| 560.128 | 560.128 | 935.492 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 5.927 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 5.927 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -161.639 |
| 544.486 | 867.766 | 560.134 | 0.000 | -0.000 | -0.000 |
| 641.865 | 770.387 | 560.134 | -0.000 | 0.000 | -0.000 |
| 560.134 | 560.134 | 935.498 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 5.927 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 5.927 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -161.639 |
| 770.353 | 641.829 | 560.099 | -0.000 | -0.000 | -0.000 |
| 754.778 | 657.406 | 560.100 | -0.000 | -0.000 | 0.000 |
| 560.100 | 560.100 | 935.463 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 5.927 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 5.927 | 0.000 |
| 0.009 | 0.001 | 0.003 | 0.000 | 0.000 | -297.118 |
| 770.415 | 641.895 | 560.163 | 0.000 | 0.000 | 0.000 |
| 867.797 | 544.507 | 560.162 | 0.000 | 0.000 | -0.000 |
| 560.162 | 560.162 | 935.526 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 5.927 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 5.927 | 0.000 |
| -0.009 | -0.001 | -0.003 | 0.000 | -0.000 | -297.118 |
| -296.514 | -98.838 | -37.890 | -0.000 | 0.000 | 0.000 |
| 334.373 | -729.725 | -37.890 | -0.000 | 0.000 | 0.000 |
| -37.890 | -37.890 | -281.498 | -0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -37.890 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -37.890 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -98.838 |
| -729.724 | 334.373 | -37.889 | 0.000 | -0.000 | -0.000 |
| -98.838 | -296.514 | -37.890 | 0.000 | -0.000 | -0.000 |
| -37.890 | -37.890 | -281.498 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -37.890 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -37.890 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -0.000 | -98.838 |
| -296.514 | -98.838 | -37.890 | -0.000 | 0.000 | 0.000 |
| -98.838 | -296.514 | -37.890 | -0.000 | -0.000 | -0.000 |
| -37.890 | -37.890 | -281.498 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -37.890 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -37.890 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -98.838 |
| -296.514 | -98.838 | -37.890 | 0.000 | 0.000 | 0.000 |
| -98.838 | -296.514 | -37.890 | 0.000 | 0.000 | 0.000 |
| -37.890 | -37.890 | -281.497 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -37.890 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -37.890 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -98.838 |
| -296.516 | -98.838 | -37.890 | -0.000 | 0.000 | -0.000 |
| -98.838 | -296.515 | -37.890 | -0.000 | -0.000 | -0.000 |
| -37.890 | -37.890 | -281.498 | -0.000 | 0.000 | 0.000 |
| 0.001 | -0.001 | -0.000 | -37.890 | 0.000 | 0.000 |
| -0.001 | 0.001 | -0.000 | -0.000 | -37.890 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -98.838 |
| -296.512 | -98.838 | -37.889 | 0.000 | 0.000 | 0.000 |
| -98.837 | -296.513 | -37.889 | 0.000 | -0.000 | 0.000 |
| -37.889 | -37.889 | -281.497 | 0.000 | 0.000 | -0.000 |
| -0.001 | 0.001 | 0.000 | -37.890 | -0.000 | -0.000 |
| 0.001 | -0.001 | 0.000 | -0.000 | -37.890 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -98.838 |
| -195.366 | -53.503 | -19.365 | -0.000 | 0.000 | 0.000 |
| -53.503 | -195.366 | -19.365 | 0.000 | 0.000 | 0.000 |
| -16.754 | -16.754 | -231.389 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -31.572 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -31.572 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -259.874 |
| -384.309 | 135.440 | -19.365 | -0.000 | 0.000 | 0.000 |
| 135.440 | -384.309 | -19.365 | -0.000 | -0.000 | 0.000 |
| -16.754 | -16.754 | -231.389 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -31.572 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -31.572 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -259.874 |
| -384.309 | 135.440 | -19.365 | 0.000 | 0.000 | -0.000 |
| -53.503 | -195.366 | -19.365 | 0.000 | 0.000 | -0.000 |
| -16.754 | -16.754 | -231.389 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -31.572 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -31.572 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -70.931 |
| -384.308 | 135.440 | -19.365 | -0.000 | 0.000 | 0.000 |
| 135.440 | -384.308 | -19.365 | -0.000 | -0.000 | 0.000 |
| -16.754 | -16.754 | -231.389 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -31.572 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -31.572 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -70.931 |
| -201.272 | -47.599 | -19.365 | -0.000 | 0.000 | -0.000 |
| -47.599 | -201.272 | -19.365 | 0.000 | 0.000 | -0.000 |
| -16.754 | -16.754 | -231.389 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -31.572 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -31.572 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -70.931 |
| -201.269 | -47.598 | -19.365 | -0.000 | -0.000 | 0.000 |
| -53.503 | -195.365 | -19.365 | -0.000 | -0.000 | 0.000 |
| -16.754 | -16.754 | -231.388 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -31.572 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -31.572 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -70.931 |
| 90.891 | 30.352 | 24.281 | 0.000 | 0.000 | 0.000 |
| 33.397 | 87.846 | 24.281 | -0.000 | 0.000 | 0.000 |
| 24.281 | 24.281 | 96.962 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 24.199 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 24.199 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 27.224 |
| 87.846 | 33.397 | 24.281 | 0.000 | -0.000 | -0.000 |
| 33.397 | 87.846 | 24.281 | 0.000 | 0.000 | -0.000 |
| 24.281 | 24.281 | 96.962 | -0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 24.199 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 24.199 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 27.224 |
| 87.846 | 33.397 | 24.281 | 0.000 | -0.000 | 0.000 |
| 30.352 | 90.891 | 24.281 | 0.000 | -0.000 | 0.000 |
| 24.281 | 24.281 | 96.962 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 24.199 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 24.199 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 30.270 |
| 90.891 | 30.352 | 24.281 | -0.000 | 0.000 | -0.000 |
| 33.397 | 87.846 | 24.281 | -0.000 | 0.000 | -0.000 |
| 24.281 | 24.281 | 96.962 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 24.199 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 24.199 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 30.270 |
| 87.846 | 33.397 | 24.281 | 0.000 | 0.000 | 0.000 |
| 33.397 | 87.846 | 24.281 | 0.000 | -0.000 | 0.000 |
| 24.281 | 24.281 | 96.962 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 24.199 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 24.199 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 27.224 |
| 87.846 | 33.397 | 24.281 | 0.000 | -0.000 | -0.000 |
| 33.397 | 87.846 | 24.281 | -0.000 | 0.000 | -0.000 |
| 24.281 | 24.281 | 96.962 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 24.199 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 24.199 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 27.224 |
| 488.603 | 662.714 | 662.714 | 0.000 | 0.000 | 0.000 |
| 662.714 | 488.603 | 662.714 | 0.000 | 0.000 | 0.000 |
| 662.714 | 662.714 | 488.603 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -289.833 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -289.833 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -289.833 |
| 488.605 | 662.715 | 662.715 | -0.000 | -0.000 | -0.000 |
| 662.715 | 488.605 | 662.715 | -0.000 | -0.000 | -0.000 |
| 662.715 | 662.715 | 488.605 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -289.833 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -289.833 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -289.833 |
| 488.596 | 662.706 | 662.706 | 0.000 | 0.000 | 0.000 |
| 662.706 | 488.596 | 662.706 | 0.000 | -0.000 | 0.000 |
| 662.706 | 662.706 | 488.596 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -289.833 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -289.833 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -289.833 |
| 488.613 | 662.723 | 662.723 | -0.000 | -0.000 | -0.000 |
| 662.723 | 488.613 | 662.723 | -0.000 | -0.000 | -0.000 |
| 662.723 | 662.723 | 488.613 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -289.833 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -289.833 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -289.833 |
| 488.518 | 662.629 | 662.629 | 0.000 | 0.000 | 0.000 |
| 662.629 | 488.518 | 662.629 | 0.000 | 0.000 | 0.000 |
| 662.629 | 662.629 | 488.518 | 0.000 | 0.000 | 0.000 |
| -0.001 | 0.000 | -0.000 | -289.833 | 0.000 | 0.000 |
| 0.000 | -0.001 | -0.000 | 0.000 | -289.833 | 0.000 |
| 0.000 | -0.000 | -0.001 | 0.000 | 0.000 | -289.833 |
| 488.691 | 662.800 | 662.800 | 0.000 | -0.000 | -0.000 |
| 662.800 | 488.691 | 662.800 | -0.000 | 0.000 | -0.000 |
| 662.800 | 662.800 | 488.691 | -0.000 | -0.000 | 0.000 |
| 0.001 | -0.000 | 0.000 | -289.833 | -0.000 | 0.000 |
| -0.000 | 0.001 | 0.000 | -0.000 | -289.833 | -0.000 |
| -0.000 | 0.000 | 0.001 | -0.000 | -0.000 | -289.833 |
| -51.864 | -30.202 | -30.202 | -0.000 | -0.000 | -0.000 |
| -30.202 | -51.864 | -30.202 | -0.000 | -0.000 | -0.000 |
| -30.202 | -30.202 | -51.864 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -30.202 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -30.202 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -30.202 |
| -51.864 | -30.202 | -30.202 | 0.000 | 0.000 | 0.000 |
| -30.202 | -51.864 | -30.202 | 0.000 | 0.000 | 0.000 |
| -30.202 | -30.202 | -51.864 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -30.202 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -30.202 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -30.202 |
| -31.768 | -14.132 | -14.132 | -0.000 | -0.000 | -0.000 |
| -14.132 | -31.768 | -14.132 | -0.000 | -0.000 | -0.000 |
| -14.132 | -14.132 | -31.768 | -0.000 | -0.000 | -0.000 |
| 0.004 | 0.004 | 0.004 | -19.436 | -0.000 | -0.000 |
| 0.004 | 0.004 | 0.004 | -0.000 | -19.436 | -0.000 |
| 0.004 | 0.004 | 0.004 | -0.000 | 0.000 | -19.436 |
| -31.777 | -14.141 | -14.141 | 0.000 | 0.000 | 0.000 |
| -14.141 | -31.777 | -14.141 | 0.000 | 0.000 | 0.000 |
| -14.141 | -14.141 | -31.777 | 0.000 | 0.000 | 0.000 |
| -0.004 | -0.004 | -0.004 | -19.436 | 0.000 | -0.000 |
| -0.004 | -0.004 | -0.004 | 0.000 | -19.436 | 0.000 |
| -0.004 | -0.004 | -0.004 | 0.000 | 0.000 | -19.436 |
| -31.768 | -14.132 | -14.132 | -0.000 | -0.000 | -0.000 |
| -14.132 | -31.768 | -14.132 | -0.000 | -0.000 | -0.000 |
| -14.132 | -14.132 | -31.768 | -0.000 | -0.000 | -0.000 |
| 0.004 | 0.004 | 0.004 | -19.436 | -0.000 | 0.000 |
| 0.004 | 0.004 | 0.004 | -0.000 | -19.436 | -0.000 |
| 0.004 | 0.004 | 0.004 | -0.000 | -0.000 | -19.436 |
| -31.777 | -14.141 | -14.141 | 0.000 | 0.000 | 0.000 |
| -14.141 | -31.777 | -14.141 | 0.000 | 0.000 | 0.000 |
| -14.141 | -14.141 | -31.777 | 0.000 | 0.000 | 0.000 |
| -0.004 | -0.004 | -0.004 | -19.436 | -0.000 | -0.000 |
| -0.004 | -0.004 | -0.004 | -0.000 | -19.436 | 0.000 |
| -0.004 | -0.004 | -0.004 | 0.000 | -0.000 | -19.436 |
| -31.768 | -14.132 | -14.132 | -0.000 | -0.000 | -0.000 |
| -14.132 | -31.768 | -14.132 | -0.000 | -0.000 | -0.000 |
| -14.132 | -14.132 | -31.768 | -0.000 | -0.000 | -0.000 |
| 0.004 | 0.004 | 0.004 | -19.436 | 0.000 | -0.000 |
| 0.004 | 0.004 | 0.004 | 0.000 | -19.436 | -0.000 |
| 0.004 | 0.004 | 0.004 | 0.000 | -0.000 | -19.436 |
| -31.776 | -14.141 | -14.141 | 0.000 | 0.000 | 0.000 |
| -14.141 | -31.776 | -14.141 | 0.000 | 0.000 | 0.000 |
| -14.141 | -14.141 | -31.776 | 0.000 | 0.000 | 0.000 |
| -0.004 | -0.004 | -0.004 | -19.436 | 0.000 | 0.000 |
| -0.004 | -0.004 | -0.004 | 0.000 | -19.436 | 0.000 |
| -0.004 | -0.004 | -0.004 | 0.000 | -0.000 | -19.436 |
| 7.907 | 7.907 | 7.907 | 0.000 | 0.000 | 0.000 |
| 7.907 | 7.907 | 7.907 | 0.000 | 0.000 | 0.000 |
| 7.907 | 7.907 | 7.907 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| 7.907 | 7.907 | 7.907 | -0.000 | -0.000 | -0.000 |
| 7.907 | 7.907 | 7.907 | -0.000 | -0.000 | -0.000 |
| 7.907 | 7.907 | 7.907 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 7.907 | 7.907 | 7.907 | 0.000 | 0.000 | 0.000 |
| 7.907 | 7.907 | 7.907 | 0.000 | 0.000 | 0.000 |
| 7.907 | 7.907 | 7.907 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| 7.907 | 7.907 | 7.907 | -0.000 | -0.000 | -0.000 |
| 7.907 | 7.907 | 7.907 | -0.000 | -0.000 | -0.000 |
| 7.907 | 7.907 | 7.907 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 7.907 | 7.907 | 7.907 | 0.000 | 0.000 | 0.000 |
| 7.907 | 7.907 | 7.907 | 0.000 | 0.000 | 0.000 |
| 7.907 | 7.907 | 7.907 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| 7.907 | 7.907 | 7.907 | -0.000 | -0.000 | -0.000 |
| 7.907 | 7.907 | 7.907 | -0.000 | -0.000 | -0.000 |
| 7.907 | 7.907 | 7.907 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 476.742 | 1390.831 | 670.189 | 0.000 | 0.000 | 0.000 |
| 1390.831 | 476.742 | 670.189 | 0.000 | -0.000 | 0.000 |
| 670.189 | 670.189 | 1001.416 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 73.130 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 73.130 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 3.800 |
| 878.477 | 989.095 | 670.189 | 0.000 | 0.000 | -0.000 |
| 989.095 | 878.477 | 670.189 | 0.000 | 0.000 | -0.000 |
| 670.189 | 670.189 | 1001.416 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 73.130 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 73.130 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 3.800 |
| 35.339 | 1832.234 | 670.184 | -0.000 | -0.000 | 0.000 |
| 1832.234 | 35.339 | 670.184 | -0.000 | 0.000 | 0.000 |
| 670.190 | 670.190 | 1001.417 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 53.923 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 53.923 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 3.800 |
| 878.476 | 989.093 | 670.188 | 0.000 | -0.000 | -0.000 |
| 989.093 | 878.476 | 670.188 | 0.000 | -0.000 | -0.000 |
| 670.187 | 670.187 | 1001.415 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 53.923 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 53.923 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 3.800 |
| 1051769.474 | -2624020.406 | 31736.516 | -930938.669 | 265977.550 | 21516.484 |
| -1734392.036 | 2021748.783 | -1001900.412 | 135328.270 | 280928.570 | 104366.443 |
| 670.204 | 670.204 | 1001.429 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 73.130 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 73.130 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 3.800 |
| 1764839.230 | -2900201.748 | -2645159.973 | 145829.919 | -39922.645 | 135831.390 |
| 1964768.878 | 3529957.402 | 3431406.638 | -298083.504 | 142141.401 | 276505.597 |
| 670.174 | 670.174 | 1001.403 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 73.130 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 73.130 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 3.800 |
| -454049282.191 | 408145108.536 | 104232856.933 | 0.000 | -0.000 | -0.000 |
| -626705548.570 | -506664120.769 | -33019378.204 | 0.000 | -0.000 | -8483575.217 |
| -6.083 | -6.083 | 54.628 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -24.712 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -24.712 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -0.000 | -0.182 |
| 41.315 | -0.168 | -6.083 | 0.000 | 0.000 | -0.000 |
| 89.257 | -48.111 | -6.083 | 0.000 | 0.000 | 0.000 |
| -6.083 | -6.083 | 54.628 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -24.712 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -24.712 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -0.182 |
| -45404874.457 | 40814513.992 | 10423299.648 | 0.000 | 0.000 | -0.000 |
| 40814513.992 | -45404874.457 | 10423299.648 | 0.000 | 0.000 | -0.000 |
| -6.083 | -6.083 | 54.628 | -0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -24.712 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -24.712 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -0.182 |
| -40814457.597 | 45404929.058 | -10423286.024 | 0.000 | 0.000 | 0.000 |
| 45404929.058 | -40814457.597 | -10423286.024 | 0.000 | -0.000 | -0.000 |
| -6.083 | -6.083 | 54.628 | -0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -24.712 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -24.712 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -0.182 |
| -4540433.683 | 4081454.537 | 1042343.919 | -0.000 | -0.000 | 0.000 |
| 4081454.537 | -4540433.683 | 1042343.919 | -0.000 | 0.000 | -0.000 |
| -6.083 | -6.083 | 54.628 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -24.712 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -24.712 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | -0.182 |
| -4081398.142 | 4540488.284 | -1042330.295 | 0.000 | -0.000 | 0.000 |
| 4540488.284 | -4081398.142 | -1042330.295 | 0.000 | -0.000 | 0.000 |
| -6.083 | -6.083 | 54.628 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -24.712 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -24.712 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | -0.182 |
| 971.590 | 595.086 | 612.688 | -0.000 | -0.000 | 0.000 |
| 595.086 | 971.590 | 612.688 | -0.000 | 0.000 | -0.000 |
| 612.688 | 612.688 | 761.780 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 102.170 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 102.170 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 17.004 |
| 971.591 | 595.086 | 612.688 | 0.000 | 0.000 | 0.000 |
| 595.086 | 971.591 | 612.688 | -0.000 | 0.000 | -0.000 |
| 612.688 | 612.688 | 761.780 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 102.170 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 102.170 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 17.004 |
| 971.591 | 595.087 | 612.688 | -0.000 | -0.000 | -0.000 |
| 595.087 | 971.591 | 612.688 | -0.000 | -0.000 | -0.000 |
| 612.688 | 612.688 | 761.780 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 102.170 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 102.170 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 17.004 |
| 971.590 | 595.086 | 612.688 | 0.000 | 0.000 | -0.000 |
| 595.086 | 971.590 | 612.688 | 0.000 | 0.000 | -0.000 |
| 612.688 | 612.688 | 761.780 | 0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 102.170 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 102.170 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 17.004 |
| 971.592 | 595.089 | 612.689 | 0.000 | -0.000 | -0.000 |
| 595.089 | 971.592 | 612.689 | 0.000 | -0.000 | -0.000 |
| 612.689 | 612.689 | 761.778 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.001 | -0.000 | 102.170 | -0.000 | -0.000 |
| -0.001 | 0.000 | -0.000 | -0.000 | 102.170 | -0.000 |
| 0.001 | 0.001 | 0.000 | -0.000 | -0.000 | 17.004 |
| 971.589 | 595.084 | 612.687 | 0.000 | 0.000 | -0.000 |
| 595.084 | 971.589 | 612.687 | -0.000 | 0.000 | 0.000 |
| 612.687 | 612.687 | 761.782 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.001 | 0.000 | 102.170 | -0.000 | -0.000 |
| 0.001 | -0.000 | 0.000 | 0.000 | 102.170 | 0.000 |
| -0.001 | -0.001 | -0.000 | -0.000 | -0.000 | 17.004 |
| -198.819 | 25.906 | -68.939 | 0.000 | 0.000 | 0.000 |
| 25.906 | -198.819 | -68.939 | 0.000 | 0.000 | 0.000 |
| -71.627 | -71.627 | -161.694 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.001 | 0.000 | -91.932 | 0.000 | 0.000 |
| 0.001 | 0.000 | 0.000 | 0.000 | -91.932 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 8.288 |
| -198.822 | 25.906 | -68.940 | 0.000 | 0.000 | -0.000 |
| 25.906 | -198.822 | -68.940 | 0.000 | 0.000 | -0.000 |
| -71.628 | -71.628 | -161.695 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.001 | -0.000 | -91.932 | -0.000 | -0.000 |
| -0.001 | -0.000 | -0.000 | -0.000 | -91.932 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 8.288 |
| -198.820 | 25.906 | -68.939 | -0.000 | -0.000 | -0.000 |
| 25.906 | -198.820 | -68.939 | -0.000 | -0.000 | 0.000 |
| -71.627 | -71.627 | -161.694 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.001 | 0.000 | -91.932 | 0.000 | 0.000 |
| 0.001 | 0.000 | 0.000 | 0.000 | -91.932 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 8.288 |
| -198.821 | 25.906 | -68.940 | 0.000 | 0.000 | -0.000 |
| 25.906 | -198.821 | -68.940 | 0.000 | 0.000 | -0.000 |
| -71.628 | -71.628 | -161.695 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.001 | -0.000 | -91.932 | -0.000 | -0.000 |
| -0.001 | -0.000 | -0.000 | -0.000 | -91.932 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 8.288 |
| -198.821 | 25.906 | -68.940 | -0.000 | -0.000 | -0.000 |
| 25.906 | -198.821 | -68.940 | -0.000 | 0.000 | 0.000 |
| -71.628 | -71.628 | -161.694 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -91.932 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -91.932 | 0.000 |
| 0.000 | 0.001 | 0.000 | 0.000 | 0.000 | 8.288 |
| -198.820 | 25.906 | -68.940 | 0.000 | 0.000 | 0.000 |
| 25.906 | -198.820 | -68.940 | 0.000 | 0.000 | 0.000 |
| -71.627 | -71.627 | -161.695 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -91.932 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -91.932 | -0.000 |
| -0.000 | -0.001 | -0.000 | -0.000 | -0.000 | 8.288 |
| 1063.075 | 873.570 | 1011.007 | -144.416 | 0.000 | 0.000 |
| 873.570 | 1063.075 | 1011.007 | 144.416 | -0.000 | -0.000 |
| 1000.984 | 1000.984 | 969.598 | -0.000 | -0.000 | -0.000 |
| -144.417 | 144.417 | -0.000 | 166.812 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 166.812 | -144.416 |
| -0.000 | 0.000 | 0.000 | 0.000 | -144.416 | 94.753 |
| 1063.077 | 873.571 | 1011.008 | -144.416 | 0.000 | -0.000 |
| 873.571 | 1063.077 | 1011.008 | 144.416 | 0.000 | -0.000 |
| 1000.986 | 1000.986 | 969.599 | 0.000 | -0.000 | -0.000 |
| -144.416 | 144.417 | -0.000 | 166.812 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 166.812 | -144.417 |
| 0.000 | -0.000 | -0.000 | -0.000 | -144.416 | 94.753 |
| 1063.071 | 873.565 | 1011.002 | -144.417 | 0.000 | 0.000 |
| 873.565 | 1063.071 | 1011.002 | 144.417 | -0.000 | 0.000 |
| 1000.979 | 1000.979 | 969.592 | 0.000 | -0.000 | -0.000 |
| -144.417 | 144.417 | 0.000 | 166.812 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 166.812 | -144.416 |
| -0.000 | 0.000 | -0.000 | -0.000 | -144.416 | 94.753 |
| 1063.082 | 873.576 | 1011.014 | -144.416 | 0.000 | -0.000 |
| 873.576 | 1063.082 | 1011.014 | 144.416 | -0.000 | -0.000 |
| 1000.991 | 1000.991 | 969.605 | -0.000 | 0.000 | -0.000 |
| -144.416 | 144.416 | -0.000 | 166.812 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 166.812 | -144.416 |
| 0.000 | -0.000 | 0.000 | 0.000 | -144.416 | 94.753 |
| 1063.021 | 873.515 | 1010.949 | -144.417 | -0.000 | 0.000 |
| 873.515 | 1063.021 | 1010.949 | 144.417 | -0.000 | -0.000 |
| 1000.923 | 1000.923 | 969.532 | -0.000 | 0.000 | 0.000 |
| -144.417 | 144.417 | 0.000 | 166.812 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 166.812 | -144.416 |
| 0.000 | -0.000 | -0.000 | -0.000 | -144.416 | 94.753 |
| 1063.132 | 873.626 | 1011.066 | -144.416 | 0.000 | -0.000 |
| 873.626 | 1063.131 | 1011.066 | 144.416 | 0.000 | -0.000 |
| 1001.046 | 1001.046 | 969.665 | 0.000 | -0.000 | -0.000 |
| -144.416 | 144.416 | -0.000 | 166.812 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 166.812 | -144.416 |
| -0.000 | 0.000 | 0.000 | 0.000 | -144.416 | 94.753 |
| 1438.214 | 814.054 | 814.054 | 0.000 | -0.000 | -0.000 |
| 814.054 | 1438.214 | 814.054 | -0.000 | -0.000 | 0.000 |
| 814.054 | 814.054 | 1438.214 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -11.850 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -11.850 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -11.850 |
| 1438.214 | 814.054 | 814.054 | 0.000 | 0.000 | 0.000 |
| 814.054 | 1438.214 | 814.054 | 0.000 | 0.000 | -0.000 |
| 814.054 | 814.054 | 1438.214 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -11.850 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -11.850 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -11.850 |
| 1438.214 | 814.054 | 814.054 | -0.000 | -0.000 | -0.000 |
| 814.054 | 1438.214 | 814.054 | -0.000 | -0.000 | 0.000 |
| 814.054 | 814.054 | 1438.214 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -11.850 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -11.850 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -11.850 |
| 1438.213 | 814.053 | 814.053 | 0.000 | 0.000 | 0.000 |
| 814.053 | 1438.213 | 814.053 | 0.000 | 0.000 | -0.000 |
| 814.053 | 814.053 | 1438.213 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -11.850 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -11.850 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -11.850 |
| 1438.216 | 814.055 | 814.055 | -0.000 | -0.000 | 0.000 |
| 814.055 | 1438.216 | 814.055 | -0.000 | -0.000 | -0.000 |
| 814.055 | 814.055 | 1438.216 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -11.850 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -11.850 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -11.850 |
| 1438.211 | 814.052 | 814.052 | -0.000 | -0.000 | -0.000 |
| 814.052 | 1438.211 | 814.052 | -0.000 | -0.000 | 0.000 |
| 814.052 | 814.052 | 1438.211 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -11.850 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -11.850 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -11.850 |
| 54.227 | -0.019 | -0.019 | 0.000 | 0.000 | -0.000 |
| -0.019 | 54.227 | -0.019 | 0.000 | 0.000 | 0.000 |
| -0.019 | -0.019 | 54.227 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.028 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.028 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -0.028 |
| 54.227 | -0.019 | -0.019 | -0.000 | -0.000 | -0.000 |
| -0.019 | 54.227 | -0.019 | -0.000 | -0.000 | -0.000 |
| -0.019 | -0.019 | 54.227 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.028 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.028 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -0.028 |
| 54.227 | -0.019 | -0.019 | 0.000 | 0.000 | 0.000 |
| -0.019 | 54.227 | -0.019 | 0.000 | 0.000 | 0.000 |
| -0.019 | -0.019 | 54.227 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.028 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -0.028 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -0.028 |
| 54.227 | -0.019 | -0.019 | -0.000 | -0.000 | -0.000 |
| -0.019 | 54.227 | -0.019 | -0.000 | -0.000 | -0.000 |
| -0.019 | -0.019 | 54.227 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.028 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.028 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -0.028 |
| 54.227 | -0.019 | -0.019 | 0.000 | 0.000 | -0.000 |
| -0.019 | 54.227 | -0.019 | 0.000 | 0.000 | 0.000 |
| -0.019 | -0.019 | 54.227 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.028 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.028 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | -0.028 |
| 54.227 | -0.019 | -0.019 | -0.000 | -0.000 | -0.000 |
| -0.019 | 54.227 | -0.019 | -0.000 | -0.000 | -0.000 |
| -0.019 | -0.019 | 54.227 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.028 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -0.028 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | -0.028 |
| -3215.086 | -3446.112 | -3446.112 | 0.000 | 0.000 | 0.000 |
| -3446.112 | -3215.086 | -3446.112 | 0.000 | 0.000 | 0.000 |
| -3446.112 | -3446.112 | -3215.086 | 0.000 | 0.000 | 0.000 |
| -4014.053 | -4014.053 | -4014.053 | 36.741 | 0.000 | 0.000 |
| -4014.053 | -4014.053 | -4014.053 | 0.000 | 36.741 | 0.000 |
| -4014.053 | -4014.053 | -4014.053 | 0.000 | -0.000 | 36.741 |
| 4813.021 | 4581.996 | 4581.996 | -0.000 | -0.000 | -0.000 |
| 4581.996 | 4813.021 | 4581.996 | -0.000 | -0.000 | -0.000 |
| 4581.996 | 4581.996 | 4813.021 | -0.000 | -0.000 | -0.000 |
| 4014.053 | 4014.053 | 4014.053 | 36.741 | 0.000 | -0.000 |
| 4014.053 | 4014.053 | 4014.053 | 0.000 | 36.741 | -0.000 |
| 4014.053 | 4014.053 | 4014.053 | 0.000 | -0.000 | 36.741 |
| 340.960 | 115.153 | 115.153 | 0.000 | 0.000 | 0.000 |
| 115.153 | 340.960 | 115.153 | 0.000 | 0.000 | 0.000 |
| 115.153 | 115.153 | 340.960 | 0.000 | 0.000 | 0.000 |
| -449.889 | -449.889 | -449.890 | 34.500 | 0.000 | 0.000 |
| -449.889 | -449.889 | -449.890 | 0.000 | 34.500 | 0.000 |
| -449.889 | -449.890 | -449.889 | -0.000 | 0.000 | 34.500 |
| 1200.383 | 969.361 | 969.361 | -0.000 | -0.000 | -0.000 |
| 969.361 | 1200.383 | 969.361 | 0.000 | -0.000 | -0.000 |
| 969.361 | 969.361 | 1200.383 | -0.000 | -0.000 | -0.000 |
| 401.200 | 401.200 | 401.200 | 36.746 | -0.000 | -0.000 |
| 401.200 | 401.200 | 401.200 | -0.000 | 36.746 | -0.000 |
| 401.200 | 401.200 | 401.200 | -0.000 | -0.000 | 36.746 |
| 758.672 | 527.655 | 527.655 | 0.000 | 0.000 | 0.000 |
| 527.655 | 758.672 | 527.655 | 0.000 | 0.000 | 0.000 |
| 527.655 | 527.655 | 758.672 | 0.000 | 0.000 | 0.000 |
| -44.936 | -44.936 | -44.936 | 34.510 | 0.000 | 0.000 |
| -44.936 | -44.936 | -44.936 | 0.000 | 34.510 | 0.000 |
| -44.936 | -44.936 | -44.936 | -0.000 | 0.000 | 34.510 |
| 842.007 | 621.806 | 621.806 | -0.000 | -0.000 | -0.000 |
| 621.806 | 842.007 | 621.806 | -0.000 | -0.000 | -0.000 |
| 621.806 | 621.806 | 842.007 | 0.000 | -0.000 | -0.000 |
| 44.936 | 44.936 | 44.936 | 34.510 | -0.000 | -0.000 |
| 44.936 | 44.936 | 44.936 | -0.000 | 34.510 | -0.000 |
| 44.936 | 44.936 | 44.936 | -0.000 | -0.000 | 34.510 |
| 1295.693 | 1072.661 | 1072.661 | 0.000 | 0.000 | 0.000 |
| 1072.661 | 1295.693 | 1072.661 | -0.000 | 0.000 | 0.000 |
| 1072.661 | 1072.661 | 1295.693 | 0.000 | 0.000 | -0.000 |
| 504.247 | 504.247 | 504.247 | 32.582 | 0.000 | -0.000 |
| 504.247 | 504.247 | 504.247 | -0.000 | 32.582 | 0.000 |
| 504.247 | 504.247 | 504.247 | 0.000 | 0.000 | 32.582 |
| 260.298 | 29.756 | 29.756 | -0.000 | -0.000 | -0.000 |
| 29.756 | 260.298 | 29.756 | 0.000 | 0.000 | 0.000 |
| 29.756 | 29.756 | 260.298 | -0.000 | -0.000 | 0.000 |
| -504.247 | -504.247 | -504.247 | 32.582 | 0.000 | 0.000 |
| -504.247 | -504.247 | -504.247 | 0.000 | 32.582 | -0.000 |
| -504.247 | -504.247 | -504.247 | 0.000 | -0.000 | 32.582 |
| 857.631 | 627.437 | 627.437 | 0.000 | 0.000 | 0.000 |
| 627.437 | 857.631 | 627.437 | 0.000 | 0.000 | -0.000 |
| 627.437 | 627.437 | 857.631 | 0.000 | 0.000 | -0.000 |
| 54.342 | 54.342 | 54.342 | 36.152 | 0.000 | 0.000 |
| 54.342 | 54.342 | 54.342 | 0.000 | 36.152 | 0.000 |
| 54.342 | 54.342 | 54.342 | 0.000 | 0.000 | 36.152 |
| 749.485 | 518.731 | 518.731 | -0.000 | -0.000 | 0.000 |
| 518.731 | 749.485 | 518.731 | -0.000 | -0.000 | 0.000 |
| 518.731 | 518.731 | 749.485 | 0.000 | -0.000 | -0.000 |
| -54.342 | -54.342 | -54.342 | 36.152 | -0.000 | 0.000 |
| -54.342 | -54.342 | -54.342 | -0.000 | 36.152 | 0.000 |
| -54.342 | -54.342 | -54.342 | 0.000 | 0.000 | 36.152 |
| 822.990 | 578.760 | 578.760 | 0.000 | 0.000 | 0.000 |
| 578.760 | 822.990 | 578.760 | 0.000 | -0.000 | 0.000 |
| 578.760 | 578.760 | 822.990 | 0.000 | 0.000 | 0.000 |
| 4.878 | 4.877 | 4.877 | 32.837 | 0.000 | 0.000 |
| 4.877 | 4.878 | 4.877 | -0.000 | 32.837 | -0.000 |
| 4.877 | 4.877 | 4.878 | 0.000 | -0.000 | 32.837 |
| 823.047 | 578.813 | 578.813 | 0.000 | 0.000 | 0.000 |
| 578.813 | 823.047 | 578.813 | 0.000 | -0.000 | -0.000 |
| 561.102 | 561.102 | 777.686 | -0.000 | -0.000 | -0.000 |
| -4.878 | -4.877 | -4.877 | 32.837 | -0.000 | 0.000 |
| -4.877 | -4.878 | -4.877 | 0.000 | 32.837 | -0.000 |
| -4.877 | -4.877 | -4.878 | 0.000 | -0.000 | 32.837 |
| 7151.546 | 7149.270 | 7149.270 | 0.000 | 0.000 | 0.000 |
| 7149.270 | 7151.546 | 7149.270 | 0.000 | 0.000 | -0.000 |
| 7149.270 | 7149.270 | 7151.546 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 91.583 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 91.583 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 91.583 |
| 591.922 | 578.365 | 578.365 | -0.000 | 0.000 | 0.000 |
| 578.365 | 591.922 | 578.365 | -0.000 | -0.000 | -0.000 |
| 578.365 | 578.365 | 591.922 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 91.583 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 91.583 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 91.583 |
| 1264.315 | 1248.136 | 1248.136 | 0.000 | 0.000 | 0.000 |
| 1248.136 | 1264.315 | 1248.136 | 0.000 | -0.000 | 0.000 |
| 1248.136 | 1248.136 | 1264.315 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 91.583 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 91.583 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 91.583 |
| -85.236 | -87.508 | -87.508 | -0.000 | -0.000 | 0.000 |
| -87.508 | -85.236 | -87.508 | 0.000 | -0.000 | -0.000 |
| -87.508 | -87.508 | -85.236 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 91.583 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 91.583 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 91.583 |
| 591.925 | 578.369 | 578.369 | 0.000 | -0.000 | 0.000 |
| 578.369 | 591.925 | 578.369 | 0.000 | 0.000 | 0.000 |
| 578.369 | 578.369 | 591.925 | 0.000 | -0.000 | 0.000 |
| 68.570 | 68.570 | 68.570 | 80.173 | 0.000 | -0.000 |
| 68.570 | 68.570 | 68.570 | 0.000 | 80.173 | 0.000 |
| 68.570 | 68.570 | 68.570 | 0.000 | -0.000 | 80.173 |
| 591.920 | 578.362 | 578.362 | 0.000 | -0.000 | -0.000 |
| 578.362 | 591.920 | 578.362 | 0.000 | 0.000 | -0.000 |
| 578.362 | 578.362 | 591.920 | -0.000 | -0.000 | -0.000 |
| -68.570 | -68.570 | -68.570 | 80.173 | -0.000 | -0.000 |
| -68.570 | -68.570 | -68.570 | 0.000 | 80.173 | -0.000 |
| -68.570 | -68.570 | -68.570 | 0.000 | -0.000 | 80.173 |
| 573.696 | 567.833 | 567.833 | 0.000 | 0.000 | 0.000 |
| 567.833 | 573.696 | 567.833 | 0.000 | 0.000 | -0.000 |
| 567.833 | 567.833 | 573.696 | 0.000 | 0.000 | -0.000 |
| 0.122 | 0.122 | 0.122 | 79.917 | 0.000 | 0.000 |
| 0.122 | 0.122 | 0.122 | 0.000 | 79.917 | 0.000 |
| 0.122 | 0.122 | 0.122 | 0.000 | 0.000 | 79.917 |
| 573.448 | 567.541 | 567.541 | -0.000 | -0.000 | -0.000 |
| 567.541 | 573.448 | 567.541 | -0.000 | 0.000 | -0.000 |
| 567.541 | 567.541 | 573.448 | -0.000 | 0.000 | -0.000 |
| -0.122 | -0.122 | -0.122 | 79.917 | -0.000 | -0.000 |
| -0.122 | -0.122 | -0.122 | -0.000 | 79.917 | -0.000 |
| -0.122 | -0.122 | -0.122 | -0.000 | -0.000 | 79.917 |
| 577.732 | 574.018 | 574.018 | 0.000 | 0.000 | 0.000 |
| 574.018 | 577.732 | 574.018 | 0.000 | 0.000 | 0.000 |
| 574.018 | 574.018 | 577.732 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 91.464 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 91.464 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 91.464 |
| 593.697 | 580.397 | 580.397 | -0.000 | 0.000 | -0.000 |
| 580.397 | 593.697 | 580.397 | -0.000 | 0.000 | -0.000 |
| 580.397 | 580.397 | 593.697 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 91.464 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 91.464 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 91.464 |
| 573.577 | 567.693 | 567.693 | 0.000 | 0.000 | 0.000 |
| 567.693 | 573.577 | 567.693 | 0.000 | 0.000 | -0.000 |
| 567.693 | 567.693 | 573.577 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 91.464 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 91.464 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 91.464 |
| 577.708 | 573.996 | 573.996 | -0.000 | -0.000 | -0.000 |
| 573.996 | 577.708 | 573.996 | -0.000 | -0.000 | -0.000 |
| 573.996 | 573.996 | 577.708 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 91.464 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 91.464 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 91.464 |
| 819.690 | 575.462 | 575.462 | 0.000 | 0.000 | -0.000 |
| 575.462 | 819.690 | 575.462 | -0.000 | 0.000 | 0.000 |
| 575.462 | 575.462 | 819.690 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 45.977 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 45.977 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 45.977 |
| 819.690 | 575.462 | 575.462 | -0.000 | -0.000 | 0.000 |
| 575.462 | 819.690 | 575.462 | -0.000 | -0.000 | 0.000 |
| 575.462 | 575.462 | 819.690 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 45.977 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 45.977 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 45.977 |
| 819.688 | 575.460 | 575.460 | 0.000 | 0.000 | 0.000 |
| 575.460 | 819.688 | 575.460 | 0.000 | 0.000 | -0.000 |
| 575.460 | 575.460 | 819.688 | -0.000 | -0.000 | 0.000 |
| -121.434 | -121.434 | -121.434 | 39.033 | 0.000 | 0.000 |
| -121.434 | -121.434 | -121.434 | 0.000 | 39.033 | 0.000 |
| -121.434 | -121.434 | -121.434 | -0.000 | 0.000 | 39.033 |
| 927.317 | 692.879 | 692.879 | -0.000 | -0.000 | 0.000 |
| 692.879 | 927.317 | 692.879 | -0.000 | -0.000 | 0.000 |
| 692.879 | 692.879 | 927.317 | 0.000 | -0.000 | -0.000 |
| 121.434 | 121.434 | 121.434 | 39.033 | -0.000 | 0.000 |
| 121.434 | 121.434 | 121.434 | 0.000 | 39.033 | -0.000 |
| 121.434 | 121.434 | 121.434 | 0.000 | -0.000 | 39.033 |
| 819.664 | 575.437 | 575.437 | 0.000 | 0.000 | 0.000 |
| 575.437 | 819.664 | 575.437 | -0.000 | 0.000 | -0.000 |
| 575.437 | 575.437 | 819.664 | 0.000 | 0.000 | -0.000 |
| -4.066 | -4.066 | -4.066 | 33.084 | -0.000 | -0.000 |
| -4.066 | -4.066 | -4.066 | -0.000 | 33.084 | -0.000 |
| -4.066 | -4.066 | -4.066 | -0.000 | 0.000 | 33.084 |
| 817.026 | 583.658 | 583.658 | -0.000 | -0.000 | 0.000 |
| 583.658 | 817.026 | 583.658 | -0.000 | -0.000 | 0.000 |
| 583.658 | 583.658 | 817.026 | -0.000 | -0.000 | -0.000 |
| 4.066 | 4.066 | 4.066 | 33.084 | -0.000 | -0.000 |
| 4.066 | 4.066 | 4.066 | -0.000 | 33.084 | 0.000 |
| 4.066 | 4.066 | 4.066 | 0.000 | -0.000 | 33.084 |
| 45.612 | 341.874 | 341.874 | 0.000 | 0.000 | 0.000 |
| 341.874 | 45.612 | 341.874 | 0.000 | 0.000 | 0.000 |
| 341.874 | 341.874 | 45.612 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -632.069 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -632.069 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | -632.069 |
| 45.612 | 341.874 | 341.874 | 0.000 | -0.000 | -0.000 |
| 341.874 | 45.612 | 341.874 | -0.000 | 0.000 | -0.000 |
| 341.874 | 341.874 | 45.612 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -632.069 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -632.069 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -632.069 |
| 45.612 | 341.874 | 341.874 | 0.000 | 0.000 | 0.000 |
| 341.874 | 45.612 | 341.874 | 0.000 | 0.000 | 0.000 |
| 341.874 | 341.874 | 45.612 | 0.000 | 0.000 | 0.000 |
| 10347254.005 | 302564073.075 | 297825874.578 | -144858576.414 | 23175.846 | -43957.708 |
| 303546514.386 | 13279169.203 | 300062425.517 | -10031.488 | -146584808.001 | -13472.845 |
| 303966984.358 | 300013471.183 | 13312911.059 | 11512.824 | -11980.056 | -144408590.065 |
| 45.613 | 341.874 | 341.874 | 0.000 | -0.000 | -0.000 |
| 341.874 | 45.613 | 341.874 | -0.000 | -0.000 | -0.000 |
| 341.874 | 341.874 | 45.613 | -0.000 | -0.000 | -0.000 |
| -32946387.792 | -332099178.735 | -325853558.934 | -172282761.897 | 47852.870 | 32466.473 |
| -349750134.022 | -42198774.039 | -342222353.395 | -13705.589 | -187196991.533 | 2568.083 |
| -321716189.988 | -313828308.091 | -22959997.546 | -20221.587 | -11455.522 | -162181150.318 |
| 45.607 | 341.870 | 341.870 | 0.000 | 0.000 | 0.000 |
| 341.870 | 45.607 | 341.870 | 0.000 | 0.000 | 0.000 |
| 341.870 | 341.870 | 45.607 | 0.000 | 0.000 | 0.000 |
| -3036989.789 | 23140962.475 | 24813380.697 | -8539702.193 | -706.961 | -914.898 |
| 23138306.395 | -3357887.826 | 24782194.491 | -370.262 | -8538579.231 | -829.043 |
| 22871017.968 | 24481255.957 | -3641615.501 | -1549.597 | -2309.147 | -8221327.786 |
| 45.618 | 341.878 | 341.878 | -0.000 | -0.000 | -0.000 |
| 341.878 | 45.618 | 341.878 | -0.000 | -0.000 | -0.000 |
| 341.878 | 341.878 | 45.618 | -0.000 | -0.000 | -0.000 |
| 5839183.242 | -18329945.298 | -19546443.475 | -5890228.327 | 652.227 | -264.243 |
| -18265017.104 | 5926935.269 | -19427402.811 | -1104.037 | -5886570.838 | -2833.791 |
| -18476595.809 | -19681288.163 | 5618108.587 | 400.623 | 462.290 | -5967095.762 |
| 0.893 | 2.764 | 2.764 | 0.000 | -0.000 | -0.000 |
| 2.764 | 0.893 | 2.764 | 0.000 | -0.000 | -0.000 |
| 2.764 | 2.764 | 0.893 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -31.794 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -31.794 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -31.794 |
| 0.892 | 2.764 | 2.764 | -0.000 | -0.000 | 0.000 |
| 2.764 | 0.892 | 2.764 | -0.000 | -0.000 | -0.000 |
| 2.764 | 2.764 | 0.892 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -31.794 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -31.794 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -31.794 |
| 0.893 | 2.764 | 2.764 | 0.000 | 0.000 | 0.000 |
| 2.764 | 0.893 | 2.764 | 0.000 | 0.000 | -0.000 |
| 2.764 | 2.764 | 0.893 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -25.405 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -25.405 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -25.405 |
| 0.893 | 2.764 | 2.764 | -0.000 | -0.000 | -0.000 |
| 2.764 | 0.893 | 2.764 | -0.000 | 0.000 | 0.000 |
| 2.764 | 2.764 | 0.893 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -25.405 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -25.405 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -25.405 |
| 0.892 | 2.764 | 2.764 | 0.000 | 0.000 | 0.000 |
| 2.764 | 0.892 | 2.764 | 0.000 | 0.000 | -0.000 |
| 2.764 | 2.764 | 0.892 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -25.405 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -25.405 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -25.405 |
| 0.893 | 2.764 | 2.764 | -0.000 | -0.000 | 0.000 |
| 2.764 | 0.893 | 2.764 | -0.000 | -0.000 | 0.000 |
| 2.764 | 2.764 | 0.893 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -25.405 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -25.405 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -25.405 |
| 40.934 | 22.104 | 22.104 | -0.000 | -0.000 | -0.000 |
| 22.104 | 40.934 | 22.104 | -0.000 | -0.000 | -0.000 |
| 22.104 | 22.104 | 40.934 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 16.820 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 16.820 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 16.820 |
| 40.934 | 22.104 | 22.104 | 0.000 | 0.000 | 0.000 |
| 22.104 | 40.934 | 22.104 | 0.000 | 0.000 | 0.000 |
| 22.104 | 22.104 | 40.934 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 16.820 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 16.820 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 16.820 |
| 40.934 | 22.104 | 22.104 | 0.000 | -0.000 | 0.000 |
| 22.104 | 40.934 | 22.104 | -0.000 | 0.000 | -0.000 |
| 22.104 | 22.104 | 40.934 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 16.820 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 16.820 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 16.820 |
| 40.934 | 22.104 | 22.104 | 0.000 | -0.000 | 0.000 |
| 22.104 | 40.934 | 22.104 | -0.000 | 0.000 | 0.000 |
| 22.104 | 22.104 | 40.934 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 16.820 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 16.820 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 16.820 |
| 40.934 | 22.104 | 22.104 | -0.000 | -0.000 | -0.000 |
| 22.104 | 40.934 | 22.104 | -0.000 | -0.000 | -0.000 |
| 22.104 | 22.104 | 40.934 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 9.893 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 9.893 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 9.893 |
| 40.933 | 22.104 | 22.104 | 0.000 | 0.000 | 0.000 |
| 22.104 | 40.933 | 22.104 | 0.000 | 0.000 | 0.000 |
| 22.104 | 22.104 | 40.933 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 9.893 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 9.893 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 9.893 |
| -2012.382 | 1668.314 | 1668.314 | 0.000 | 0.000 | 0.000 |
| 1668.314 | -2012.382 | 1668.314 | 0.000 | 0.000 | 0.000 |
| 1668.314 | 1668.314 | -2012.382 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -670.411 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -670.411 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | -670.411 |
| 686.777 | 318.732 | 318.732 | -0.000 | 0.000 | -0.000 |
| 318.732 | 686.777 | 318.732 | -0.000 | -0.000 | -0.000 |
| 318.732 | 318.732 | 686.777 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -670.411 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -670.411 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -670.411 |
| -2012.372 | 1668.326 | 1668.326 | 0.000 | 0.000 | 0.000 |
| 1668.326 | -2012.372 | 1668.326 | 0.000 | 0.000 | -0.000 |
| 1668.326 | 1668.326 | -2012.372 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 42.259 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 42.259 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 42.259 |
| 686.777 | 318.731 | 318.731 | -0.000 | -0.000 | -0.000 |
| 318.731 | 686.777 | 318.731 | -0.000 | -0.000 | -0.000 |
| 318.731 | 318.731 | 686.777 | -0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 42.259 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 42.259 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 42.259 |
| -3543.058 | 2434.074 | 2434.074 | 0.000 | -0.000 | -0.000 |
| 2434.074 | -3543.058 | 2434.074 | -0.000 | 0.000 | -0.000 |
| 2434.074 | 2434.074 | -3543.058 | -0.000 | 0.000 | 0.000 |
| -0.001 | 0.008 | 0.007 | -670.411 | 0.000 | 0.000 |
| 0.008 | -0.001 | 0.007 | -0.000 | -670.411 | 0.000 |
| 0.008 | 0.007 | -0.001 | 0.000 | 0.000 | -670.411 |
| -3325.828 | 2324.652 | 2324.652 | -0.000 | -0.000 | 0.000 |
| 2324.652 | -3325.828 | 2324.652 | -0.000 | -0.000 | -0.000 |
| 2324.652 | 2324.652 | -3325.828 | -0.000 | -0.000 | -0.000 |
| 0.001 | -0.008 | -0.007 | -670.411 | -0.000 | -0.000 |
| -0.008 | 0.001 | -0.007 | -0.000 | -670.411 | -0.000 |
| -0.008 | -0.007 | 0.001 | -0.000 | -0.000 | -670.411 |
| -835.628 | -25.944 | -25.943 | -0.000 | -0.000 | 0.000 |
| -25.943 | -835.628 | -25.944 | -0.000 | 0.000 | -0.000 |
| -25.944 | -25.943 | -835.628 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -190.402 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -190.402 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -190.402 |
| -506.711 | -190.402 | -190.402 | -0.000 | -0.000 | -0.000 |
| -190.402 | -506.711 | -190.402 | 0.000 | -0.000 | 0.000 |
| -190.402 | -190.402 | -506.711 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -190.402 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -190.402 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -190.402 |
| -506.712 | -190.402 | -190.402 | 0.000 | 0.000 | 0.000 |
| -190.402 | -506.712 | -190.402 | -0.000 | 0.000 | 0.000 |
| -190.402 | -190.402 | -506.712 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -190.402 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -190.402 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -190.402 |
| -506.711 | -190.402 | -190.402 | -0.000 | -0.000 | -0.000 |
| -190.402 | -506.711 | -190.402 | 0.000 | -0.000 | 0.000 |
| -190.402 | -190.402 | -506.711 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -190.402 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -190.402 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -190.402 |
| -506.713 | -190.402 | -190.402 | -0.000 | 0.000 | 0.000 |
| -190.402 | -506.713 | -190.402 | -0.000 | 0.000 | 0.000 |
| -190.402 | -190.402 | -506.713 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -190.402 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -190.402 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -190.402 |
| -506.710 | -190.401 | -190.401 | -0.000 | -0.000 | -0.000 |
| -190.401 | -506.710 | -190.401 | -0.000 | -0.000 | 0.000 |
| -190.401 | -190.401 | -506.710 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -190.402 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -190.402 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -190.402 |
| -371.339 | -36.904 | -36.904 | -0.000 | -0.000 | -0.000 |
| -36.904 | -371.339 | -36.904 | -0.000 | -0.000 | 0.000 |
| -36.904 | -36.903 | -371.339 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -108.089 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -108.088 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -108.088 |
| -464.991 | 9.922 | 9.922 | -0.000 | -0.000 | 0.000 |
| 9.922 | -464.991 | 9.922 | 0.000 | -0.000 | -0.000 |
| 9.922 | 9.922 | -464.991 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -108.088 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -108.088 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -108.088 |
| -371.339 | -36.904 | -36.904 | -0.000 | 0.000 | 0.000 |
| -36.904 | -371.339 | -36.904 | 0.000 | -0.000 | -0.000 |
| -36.904 | -36.904 | -371.339 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -108.088 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -108.088 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -108.088 |
| -371.339 | -36.904 | -36.904 | 0.000 | 0.000 | 0.000 |
| -36.904 | -371.339 | -36.904 | 0.000 | 0.000 | 0.000 |
| -36.904 | -36.904 | -371.339 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -108.088 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -108.088 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -108.088 |
| -371.340 | -36.904 | -36.904 | -0.000 | -0.000 | -0.000 |
| -36.904 | -371.340 | -36.904 | -0.000 | 0.000 | 0.000 |
| -36.904 | -36.904 | -371.340 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -108.088 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -108.088 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -108.088 |
| -371.338 | -36.903 | -36.903 | 0.000 | 0.000 | -0.000 |
| -36.903 | -371.338 | -36.903 | 0.000 | 0.000 | -0.000 |
| -36.903 | -36.903 | -371.338 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -108.088 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -108.088 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -108.088 |
| 84.517 | 28.123 | 28.123 | -0.000 | -0.000 | -0.000 |
| 28.123 | 84.517 | 28.123 | -0.000 | 0.000 | -0.000 |
| 28.123 | 28.123 | 84.517 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 25.512 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 25.512 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 25.512 |
| 84.509 | 28.127 | 28.127 | 0.000 | 0.000 | 0.000 |
| 28.127 | 84.509 | 28.127 | -0.000 | 0.000 | 0.000 |
| 28.127 | 28.127 | 84.509 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 25.512 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 25.512 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 25.512 |
| 84.509 | 28.127 | 28.127 | 0.000 | -0.000 | -0.000 |
| 28.127 | 84.509 | 28.127 | 0.000 | -0.000 | -0.000 |
| 28.127 | 28.127 | 84.509 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 25.512 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 25.512 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 25.512 |
| 84.509 | 28.127 | 28.127 | -0.000 | 0.000 | 0.000 |
| 28.127 | 84.509 | 28.127 | -0.000 | 0.000 | 0.000 |
| 28.127 | 28.127 | 84.509 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 25.512 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 25.512 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 25.512 |
| 84.509 | 28.127 | 28.127 | 0.000 | -0.000 | -0.000 |
| 28.127 | 84.509 | 28.127 | 0.000 | -0.000 | -0.000 |
| 28.127 | 28.127 | 84.509 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 25.512 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 25.512 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 25.512 |
| 84.509 | 28.127 | 28.127 | -0.000 | 0.000 | 0.000 |
| 28.127 | 84.509 | 28.127 | -0.000 | 0.000 | 0.000 |
| 28.127 | 28.127 | 84.509 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 25.512 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 25.512 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 25.512 |
| 808.381 | 574.826 | 574.826 | 0.000 | 0.000 | 0.000 |
| 574.826 | 808.381 | 574.826 | 0.000 | 0.000 | 0.000 |
| 574.826 | 574.826 | 808.381 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 277.105 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 277.105 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 277.105 |
| 808.381 | 574.826 | 574.826 | -0.000 | -0.000 | -0.000 |
| 574.826 | 808.381 | 574.826 | -0.000 | -0.000 | -0.000 |
| 574.826 | 574.826 | 808.381 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 277.105 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 277.105 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 277.105 |
| 808.380 | 574.826 | 574.826 | 0.000 | 0.000 | 0.000 |
| 574.826 | 808.380 | 574.826 | 0.000 | 0.000 | 0.000 |
| 574.826 | 574.826 | 808.380 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -431.881 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -431.881 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -431.881 |
| 808.382 | 574.827 | 574.827 | -0.000 | -0.000 | -0.000 |
| 574.827 | 808.382 | 574.827 | -0.000 | -0.000 | -0.000 |
| 574.827 | 574.827 | 808.382 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -431.881 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -431.881 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -431.881 |
| 808.374 | 574.821 | 574.821 | 0.000 | 0.000 | 0.000 |
| 574.821 | 808.374 | 574.821 | 0.000 | 0.000 | -0.000 |
| 574.821 | 574.821 | 808.374 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.001 | -0.001 | 277.105 | 0.000 | 0.000 |
| -0.001 | -0.000 | -0.001 | -0.000 | 277.105 | 0.000 |
| -0.001 | -0.001 | -0.000 | 0.000 | 0.000 | 277.105 |
| 808.388 | 574.832 | 574.832 | -0.000 | -0.000 | -0.000 |
| 574.832 | 808.388 | 574.832 | -0.000 | -0.000 | -0.000 |
| 574.832 | 574.832 | 808.388 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.001 | 0.001 | 277.105 | 0.000 | -0.000 |
| 0.001 | 0.000 | 0.001 | 0.000 | 277.105 | -0.000 |
| 0.001 | 0.001 | 0.000 | -0.000 | -0.000 | 277.105 |
| -377.670 | -222.590 | -222.590 | -0.000 | -0.000 | -0.000 |
| -222.590 | -377.670 | -222.590 | -0.000 | -0.000 | 0.000 |
| -222.590 | -222.590 | -377.670 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -222.590 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -222.590 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -222.590 |
| -377.670 | -222.590 | -222.590 | 0.000 | -0.000 | -0.000 |
| -222.590 | -377.670 | -222.590 | -0.000 | 0.000 | 0.000 |
| -222.590 | -222.590 | -377.670 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -222.590 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -222.590 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -222.590 |
| -377.671 | -222.590 | -222.590 | 0.000 | 0.000 | 0.000 |
| -222.590 | -377.671 | -222.590 | -0.000 | -0.000 | 0.000 |
| -222.590 | -222.590 | -377.671 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -222.590 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -222.590 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -222.590 |
| -377.670 | -222.589 | -222.589 | -0.000 | 0.000 | -0.000 |
| -222.589 | -377.670 | -222.589 | -0.000 | -0.000 | -0.000 |
| -222.589 | -222.589 | -377.670 | -0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -222.590 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -222.590 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -222.590 |
| -377.673 | -222.590 | -222.590 | 0.000 | -0.000 | -0.000 |
| -222.590 | -377.673 | -222.590 | 0.000 | -0.000 | -0.000 |
| -222.590 | -222.590 | -377.673 | 0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -222.590 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -222.590 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -222.590 |
| -377.667 | -222.589 | -222.589 | -0.000 | 0.000 | -0.000 |
| -222.589 | -377.667 | -222.589 | 0.000 | -0.000 | -0.000 |
| -222.589 | -222.589 | -377.667 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -222.590 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -222.590 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -222.590 |
| -149.135 | -111.184 | -111.184 | 0.000 | 0.000 | 0.000 |
| -111.184 | -149.135 | -111.184 | 0.000 | 0.000 | 0.000 |
| -111.183 | -111.183 | -149.135 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -309.345 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -309.345 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -309.345 |
| -149.135 | -111.184 | -111.184 | -0.000 | -0.000 | -0.000 |
| -111.184 | -149.135 | -111.184 | -0.000 | -0.000 | -0.000 |
| -111.184 | -111.184 | -149.135 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -309.345 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -309.345 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -309.345 |
| -149.135 | -111.184 | -111.184 | 0.000 | 0.000 | 0.000 |
| -111.184 | -149.135 | -111.184 | 0.000 | 0.000 | 0.000 |
| -111.184 | -111.184 | -149.135 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -309.345 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -309.345 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -178.551 |
| -149.135 | -111.184 | -111.184 | -0.000 | 0.000 | -0.000 |
| -111.184 | -149.135 | -111.184 | -0.000 | -0.000 | -0.000 |
| -111.184 | -111.184 | -149.135 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -309.345 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -309.345 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -309.345 |
| -149.136 | -111.184 | -111.184 | 0.000 | 0.000 | 0.000 |
| -111.184 | -149.136 | -111.184 | 0.000 | 0.000 | 0.000 |
| -111.184 | -111.184 | -149.136 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -178.551 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -178.551 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -178.551 |
| -149.134 | -111.183 | -111.183 | -0.000 | -0.000 | -0.000 |
| -111.183 | -149.134 | -111.183 | -0.000 | -0.000 | -0.000 |
| -111.183 | -111.183 | -149.134 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -178.551 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -178.551 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -178.551 |
| 44.904 | 31.555 | 31.555 | 0.000 | 0.000 | 0.000 |
| 31.555 | 44.904 | 31.555 | 0.000 | 0.000 | 0.000 |
| 31.555 | 31.555 | 44.904 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 30.616 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 30.616 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 30.616 |
| 44.904 | 31.555 | 31.555 | -0.000 | -0.000 | -0.000 |
| 31.555 | 44.904 | 31.555 | -0.000 | -0.000 | -0.000 |
| 31.555 | 31.555 | 44.904 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 30.616 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 30.616 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 30.616 |
| 44.904 | 31.555 | 31.555 | 0.000 | 0.000 | -0.000 |
| 31.555 | 44.904 | 31.555 | 0.000 | 0.000 | 0.000 |
| 31.555 | 31.555 | 44.904 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 30.616 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 30.616 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 30.616 |
| 44.903 | 31.555 | 31.555 | -0.000 | -0.000 | 0.000 |
| 31.555 | 44.903 | 31.555 | -0.000 | -0.000 | -0.000 |
| 31.555 | 31.555 | 44.903 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 30.616 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 30.616 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 30.616 |
| 44.904 | 31.555 | 31.555 | 0.000 | 0.000 | 0.000 |
| 31.555 | 44.904 | 31.555 | 0.000 | 0.000 | 0.000 |
| 31.555 | 31.555 | 44.904 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 30.616 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 30.616 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 30.616 |
| 44.903 | 31.555 | 31.555 | -0.000 | -0.000 | -0.000 |
| 31.555 | 44.903 | 31.555 | -0.000 | -0.000 | -0.000 |
| 31.555 | 31.555 | 44.903 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 30.616 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 30.616 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 30.616 |
| 431.829 | 562.797 | 562.797 | 0.000 | 0.000 | 0.000 |
| 562.797 | 431.829 | 562.797 | 0.000 | 0.000 | 0.000 |
| 562.797 | 562.797 | 431.829 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 265.719 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 265.719 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 265.719 |
| 431.829 | 562.797 | 562.797 | -0.000 | -0.000 | 0.000 |
| 562.797 | 431.829 | 562.797 | -0.000 | -0.000 | -0.000 |
| 562.797 | 562.797 | 431.829 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 265.719 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 265.719 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 265.719 |
| 431.829 | 562.798 | 562.798 | 0.000 | 0.000 | 0.000 |
| 562.798 | 431.829 | 562.798 | 0.000 | 0.000 | 0.000 |
| 562.798 | 562.798 | 431.829 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 265.719 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 265.719 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 265.719 |
| 431.828 | 562.796 | 562.796 | -0.000 | -0.000 | -0.000 |
| 562.796 | 431.828 | 562.796 | -0.000 | -0.000 | -0.000 |
| 562.796 | 562.796 | 431.828 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 265.719 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 265.719 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 265.719 |
| 431.836 | 562.804 | 562.804 | 0.000 | 0.000 | 0.000 |
| 562.804 | 431.836 | 562.804 | 0.000 | 0.000 | 0.000 |
| 562.804 | 562.804 | 431.836 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.001 | 265.719 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.001 | 0.000 | 265.719 | 0.000 |
| 0.000 | 0.001 | -0.000 | 0.000 | 0.000 | 265.719 |
| 431.821 | 562.790 | 562.790 | -0.000 | -0.000 | 0.000 |
| 562.790 | 431.821 | 562.790 | -0.000 | -0.000 | -0.000 |
| 562.790 | 562.790 | 431.821 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.001 | 265.719 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.001 | -0.000 | 265.719 | -0.000 |
| -0.000 | -0.001 | 0.000 | -0.000 | -0.000 | 265.719 |
| -130.807 | 21.695 | 21.695 | -0.000 | -0.000 | -0.000 |
| 21.695 | -130.807 | 21.695 | -0.000 | -0.000 | -0.000 |
| 21.695 | 21.695 | -130.807 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -46.190 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -46.190 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -46.190 |
| -130.807 | 21.695 | 21.695 | 0.000 | -0.000 | 0.000 |
| 21.695 | -130.807 | 21.695 | -0.000 | 0.000 | 0.000 |
| 21.695 | 21.695 | -130.807 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -46.190 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -46.190 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -46.190 |
| -130.807 | 21.695 | 21.695 | -0.000 | 0.000 | -0.000 |
| 21.695 | -130.807 | 21.695 | 0.000 | 0.000 | 0.000 |
| 21.695 | 21.695 | -130.807 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -46.190 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -46.190 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -46.190 |
| -130.807 | 21.695 | 21.695 | 0.000 | -0.000 | 0.000 |
| 21.695 | -130.807 | 21.695 | 0.000 | -0.000 | 0.000 |
| 21.695 | 21.695 | -130.807 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -46.190 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -46.190 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | -46.190 |
| -130.808 | 21.695 | 21.695 | 0.000 | 0.000 | -0.000 |
| 21.695 | -130.808 | 21.695 | 0.000 | 0.000 | -0.000 |
| 21.695 | 21.695 | -130.808 | -0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -46.190 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -46.190 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | -46.190 |
| -130.807 | 21.696 | 21.696 | 0.000 | -0.000 | 0.000 |
| 21.696 | -130.807 | 21.696 | 0.000 | -0.000 | 0.000 |
| 21.696 | 21.696 | -130.807 | -0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -46.190 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -46.190 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -46.190 |
| 48.471 | 32.543 | 32.543 | -0.000 | -0.000 | 0.000 |
| 32.543 | 48.471 | 32.543 | -0.000 | 0.000 | -0.000 |
| 32.543 | 32.543 | 48.471 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 32.277 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 32.277 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 32.277 |
| 48.471 | 32.543 | 32.543 | 0.000 | 0.000 | -0.000 |
| 32.543 | 48.471 | 32.543 | 0.000 | 0.000 | -0.000 |
| 32.543 | 32.543 | 48.471 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 32.277 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 32.277 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 32.277 |
| 48.471 | 32.543 | 32.543 | -0.000 | -0.000 | 0.000 |
| 32.543 | 48.471 | 32.543 | -0.000 | 0.000 | 0.000 |
| 32.543 | 32.543 | 48.471 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 32.277 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 32.277 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 32.277 |
| 48.471 | 32.543 | 32.543 | -0.000 | 0.000 | -0.000 |
| 32.543 | 48.471 | 32.543 | 0.000 | -0.000 | -0.000 |
| 32.543 | 32.543 | 48.471 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 32.277 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 32.277 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 32.277 |
| 48.470 | 32.543 | 32.543 | -0.000 | -0.000 | 0.000 |
| 32.543 | 48.470 | 32.543 | -0.000 | -0.000 | 0.000 |
| 32.543 | 32.543 | 48.470 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 32.277 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 32.277 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 32.277 |
| 48.471 | 32.543 | 32.543 | -0.000 | 0.000 | -0.000 |
| 32.543 | 48.471 | 32.543 | 0.000 | 0.000 | -0.000 |
| 32.543 | 32.543 | 48.471 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 32.277 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 32.277 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 32.277 |
| 34.621 | 688.440 | 504.766 | 0.000 | 0.000 | 0.000 |
| 688.440 | 34.621 | 504.766 | 0.000 | -0.000 | 0.000 |
| 504.766 | 504.766 | 374.568 | 0.000 | 0.000 | 0.000 |
| 0.001 | 0.001 | 0.001 | -1929.750 | 0.000 | 0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | -1929.750 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 178.544 |
| 34.621 | 688.440 | 504.766 | -0.000 | -0.000 | 0.000 |
| 688.440 | 34.621 | 504.766 | -0.000 | 0.000 | 0.000 |
| 504.766 | 504.766 | 374.568 | -0.000 | 0.000 | -0.000 |
| -0.001 | -0.001 | -0.001 | -1929.750 | 0.000 | -0.000 |
| -0.001 | -0.001 | -0.001 | -0.000 | -1929.750 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 178.544 |
| 255.216 | 467.843 | 504.765 | 0.000 | 0.000 | 0.000 |
| 467.843 | 255.216 | 504.765 | 0.000 | 0.000 | 0.000 |
| 504.765 | 504.765 | 374.567 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 227.607 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 227.607 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 178.544 |
| 34.622 | 688.440 | 504.766 | -0.000 | 0.000 | -0.000 |
| 688.440 | 34.622 | 504.766 | 0.000 | -0.000 | -0.000 |
| 504.766 | 504.766 | 374.569 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 227.607 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 227.607 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 178.544 |
| 34.614 | 688.432 | 504.758 | 0.000 | 0.000 | 0.000 |
| 688.432 | 34.614 | 504.758 | -0.000 | 0.000 | 0.000 |
| 504.757 | 504.757 | 374.560 | 0.000 | 0.000 | 0.000 |
| 8290596.032 | 8267171.550 | 8814879.381 | 9068.328 | -172500.889 | 48098.261 |
| 8464197.616 | 8325338.647 | 9085886.887 | 19999.625 | 230794.904 | -369107.169 |
| 0.000 | 0.001 | -0.000 | 0.000 | 0.000 | 178.544 |
| 34.628 | 688.447 | 504.773 | -0.000 | -0.000 | -0.000 |
| 688.447 | 34.628 | 504.773 | -0.000 | -0.000 | -0.000 |
| 504.774 | 504.774 | 374.576 | -0.000 | -0.000 | -0.000 |
| -8303982.112 | -8204731.648 | -8710837.858 | -118246.456 | 176240.029 | 101754.179 |
| -8362443.233 | -8614885.451 | -8598144.008 | -269978.818 | 21559.460 | 144380.544 |
| -0.000 | -0.001 | 0.000 | -0.000 | -0.000 | 178.544 |
| 586.575 | 405.805 | 503.692 | 0.000 | 0.000 | -0.000 |
| 405.808 | 745.882 | 323.465 | 0.000 | 0.000 | 0.000 |
| 503.694 | 323.465 | 1169.663 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 43.184 | 0.000 | -0.000 |
| -227.524 | 11.812 | -56.782 | 0.000 | 207.995 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 132.931 |
| 586.574 | 405.805 | 503.692 | -0.000 | 0.000 | 0.000 |
| 405.807 | 745.882 | 323.465 | -0.000 | -0.000 | 0.000 |
| 503.694 | 323.464 | 1169.662 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 43.185 | -0.000 | 0.000 |
| 227.524 | -11.812 | 56.782 | -0.000 | 207.995 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 132.931 |
| 586.575 | 405.806 | 503.692 | 0.000 | -0.000 | -0.000 |
| 405.808 | 745.883 | 323.465 | 0.000 | 0.000 | -0.000 |
| 503.694 | 323.465 | 1169.663 | 0.000 | 0.000 | -0.000 |
| -23.942 | -3.792 | -10.788 | 43.021 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 208.799 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 132.931 |
| 551.595 | 391.999 | 480.031 | -0.000 | 0.000 | 0.000 |
| 400.903 | 732.556 | 309.261 | -0.000 | -0.000 | 0.000 |
| 503.693 | 323.464 | 1169.661 | -0.000 | -0.000 | 0.000 |
| 23.942 | 3.792 | 10.788 | 43.021 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 208.799 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 132.931 |
| 586.582 | 405.808 | 503.697 | 0.000 | 0.000 | -0.000 |
| 387.929 | 737.074 | 309.692 | -0.000 | -0.000 | -0.000 |
| 472.326 | 310.697 | 1148.076 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 43.185 | -0.000 | 0.000 |
| 0.001 | 0.000 | 0.001 | 0.000 | 208.799 | -0.000 |
| -1.828 | 0.249 | -0.310 | -0.000 | 0.000 | 128.766 |
| 586.568 | 405.802 | 503.687 | -0.000 | -0.000 | 0.000 |
| 389.753 | 736.953 | 310.133 | -0.000 | 0.000 | 0.000 |
| 503.687 | 323.460 | 1169.653 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 43.185 | 0.000 | 0.000 |
| -0.001 | -0.000 | -0.001 | 0.000 | 208.799 | 0.000 |
| 1.828 | -0.249 | 0.310 | 0.000 | 0.000 | 128.766 |
| 202.935 | -41.566 | 677.537 | -0.000 | -0.000 | 0.000 |
| 311.775 | 745.003 | 346.712 | -0.000 | 0.000 | -0.000 |
| 432.227 | 348.970 | 266.478 | -0.000 | -0.000 | 0.000 |
| -195.551 | -308.870 | 346.764 | 88.429 | -0.000 | -0.000 |
| -197.074 | -311.274 | 349.463 | -0.000 | 162.208 | -0.000 |
| -194.125 | -306.617 | 344.234 | -0.000 | 0.000 | 52.989 |
| 436.687 | 322.495 | 440.689 | 0.000 | 0.000 | 0.000 |
| 417.382 | 923.875 | -257.113 | 0.000 | 0.000 | 0.000 |
| 417.066 | 355.904 | -772.639 | -0.000 | 0.000 | 0.000 |
| 195.551 | 308.870 | -346.764 | 88.429 | 0.000 | -0.000 |
| 197.074 | 311.274 | -349.463 | -0.000 | 162.208 | 0.000 |
| 194.125 | 306.617 | -344.234 | 0.000 | 0.000 | 52.989 |
| 382.377 | 240.065 | 421.289 | 0.000 | 0.000 | -0.000 |
| 311.775 | 745.004 | 346.712 | -0.000 | -0.000 | -0.000 |
| 373.231 | 264.196 | 80.803 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 109.621 | -0.000 | -0.000 |
| -23.491 | -37.104 | 41.656 | 0.000 | 157.487 | -0.000 |
| -6.688 | -12.382 | 74.594 | -0.000 | -0.000 | 68.942 |
| 436.687 | 322.494 | 440.688 | 0.000 | 0.000 | -0.000 |
| 311.774 | 745.003 | 346.711 | 0.000 | 0.000 | -0.000 |
| 355.342 | 241.231 | -70.071 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 109.621 | 0.000 | 0.000 |
| 23.491 | 37.104 | -41.656 | 0.000 | 157.487 | 0.000 |
| 6.688 | 12.382 | -74.594 | 0.000 | 0.000 | 68.942 |
| 392.763 | 256.741 | 393.439 | -0.000 | -0.000 | -0.000 |
| 247.920 | 648.939 | 294.503 | -0.000 | 0.000 | -0.000 |
| 376.798 | 271.669 | 10.918 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.001 | 0.001 | 109.621 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.001 | -0.000 | 186.798 | -0.000 |
| -0.931 | -1.471 | 1.654 | 0.000 | -0.000 | 68.254 |
| 394.991 | 259.938 | 400.681 | -0.000 | -0.000 | -0.000 |
| 311.771 | 744.998 | 346.707 | -0.000 | 0.000 | -0.000 |
| 432.218 | 348.964 | 266.467 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.001 | -0.001 | 109.621 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.001 | 0.000 | 186.798 | 0.000 |
| 0.931 | 1.471 | -1.654 | 0.000 | 0.000 | 68.254 |
| 56.521 | 148.746 | 148.746 | -0.000 | -0.000 | -0.000 |
| 148.746 | 56.521 | 148.746 | -0.000 | 0.000 | -0.000 |
| 148.746 | 148.746 | 56.521 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 101.673 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 101.673 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 101.673 |
| 56.521 | 148.746 | 148.746 | -0.000 | -0.000 | 0.000 |
| 148.746 | 56.521 | 148.746 | -0.000 | 0.000 | 0.000 |
| 148.746 | 148.746 | 56.521 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 101.673 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 101.673 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 101.673 |
| 56.521 | 148.746 | 148.746 | -0.000 | -0.000 | 0.000 |
| 148.746 | 56.521 | 148.746 | 0.000 | -0.000 | -0.000 |
| 148.746 | 148.746 | 56.521 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 101.673 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 101.673 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 101.673 |
| 56.522 | 148.746 | 148.746 | -0.000 | 0.000 | -0.000 |
| 148.746 | 56.522 | 148.746 | 0.000 | 0.000 | -0.000 |
| 148.746 | 148.746 | 56.522 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 101.673 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 101.673 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 101.673 |
| 56.521 | 148.746 | 148.746 | -0.000 | -0.000 | 0.000 |
| 148.746 | 56.521 | 148.746 | 0.000 | -0.000 | -0.000 |
| 148.746 | 148.746 | 56.521 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 101.673 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 101.673 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 101.673 |
| 56.522 | 148.746 | 148.746 | 0.000 | -0.000 | -0.000 |
| 148.746 | 56.522 | 148.746 | -0.000 | 0.000 | 0.000 |
| 148.746 | 148.746 | 56.522 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 101.673 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 101.673 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 101.673 |
| -249.824 | -50.042 | -50.042 | -0.000 | 0.000 | -0.000 |
| -50.042 | -249.824 | -50.042 | 0.000 | -0.000 | -0.000 |
| -50.042 | -50.042 | -249.824 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -26.869 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -26.869 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -26.869 |
| -249.824 | -50.042 | -50.042 | -0.000 | 0.000 | 0.000 |
| -50.042 | -249.824 | -50.042 | -0.000 | 0.000 | 0.000 |
| -50.042 | -50.042 | -249.824 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -26.869 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -26.869 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -26.869 |
| -249.824 | -50.042 | -50.042 | -0.000 | 0.000 | 0.000 |
| -50.042 | -249.824 | -50.042 | 0.000 | -0.000 | -0.000 |
| -50.042 | -50.042 | -249.824 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -26.869 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -26.869 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -26.869 |
| -249.824 | -50.042 | -50.042 | 0.000 | -0.000 | 0.000 |
| -50.042 | -249.824 | -50.042 | -0.000 | 0.000 | -0.000 |
| -50.042 | -50.042 | -249.824 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -26.869 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -26.869 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | -26.869 |
| -249.825 | -50.043 | -50.043 | -0.000 | -0.000 | -0.000 |
| -50.043 | -249.825 | -50.043 | -0.000 | -0.000 | 0.000 |
| -50.043 | -50.043 | -249.825 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -26.869 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -26.869 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | -26.869 |
| -249.823 | -50.042 | -50.042 | -0.000 | -0.000 | 0.000 |
| -50.042 | -249.823 | -50.042 | -0.000 | -0.000 | 0.000 |
| -50.042 | -50.042 | -249.823 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -26.869 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -26.869 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -26.869 |
| -188.996 | -29.771 | -29.771 | 0.000 | 0.000 | -0.000 |
| -29.771 | -188.996 | -29.771 | 0.000 | 0.000 | -0.000 |
| -29.771 | -29.771 | -188.996 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -30.687 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -30.687 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | -30.687 |
| -188.996 | -29.771 | -29.771 | -0.000 | -0.000 | 0.000 |
| -29.771 | -188.996 | -29.771 | -0.000 | -0.000 | 0.000 |
| -29.771 | -29.771 | -188.996 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -30.687 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -30.687 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -30.687 |
| -188.996 | -29.771 | -29.771 | 0.000 | 0.000 | -0.000 |
| -29.771 | -188.996 | -29.771 | -0.000 | 0.000 | -0.000 |
| -29.771 | -29.771 | -188.996 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -30.687 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -30.687 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | -30.687 |
| -188.996 | -29.771 | -29.771 | 0.000 | -0.000 | -0.000 |
| -29.771 | -188.996 | -29.771 | -0.000 | -0.000 | 0.000 |
| -29.771 | -29.771 | -188.996 | -0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -30.687 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -30.687 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -30.687 |
| -188.997 | -29.771 | -29.771 | -0.000 | 0.000 | 0.000 |
| -29.771 | -188.997 | -29.771 | 0.000 | 0.000 | -0.000 |
| -29.771 | -29.771 | -188.997 | 0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -30.687 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -30.687 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | -30.687 |
| -188.996 | -29.771 | -29.771 | -0.000 | 0.000 | -0.000 |
| -29.771 | -188.996 | -29.771 | -0.000 | -0.000 | 0.000 |
| -29.771 | -29.771 | -188.996 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -30.687 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -30.687 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | -30.687 |
| 76.092 | 10.711 | 10.711 | 0.000 | 0.000 | -0.000 |
| 10.711 | 76.092 | 10.711 | 0.000 | 0.000 | -0.000 |
| 10.711 | 10.711 | 76.092 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 10.089 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 10.089 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 10.089 |
| 76.092 | 10.711 | 10.711 | -0.000 | -0.000 | 0.000 |
| 10.711 | 76.092 | 10.711 | -0.000 | -0.000 | 0.000 |
| 10.711 | 10.711 | 76.092 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 10.089 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 10.089 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 10.089 |
| 76.092 | 10.711 | 10.711 | 0.000 | 0.000 | -0.000 |
| 10.711 | 76.092 | 10.711 | 0.000 | 0.000 | -0.000 |
| 10.711 | 10.711 | 76.092 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 10.089 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 10.089 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 10.089 |
| 76.092 | 10.711 | 10.711 | -0.000 | 0.000 | -0.000 |
| 10.711 | 76.092 | 10.711 | 0.000 | 0.000 | -0.000 |
| 10.711 | 10.711 | 76.092 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 10.089 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 10.089 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 10.089 |
| 76.093 | 10.711 | 10.711 | 0.000 | -0.000 | -0.000 |
| 10.711 | 76.093 | 10.711 | 0.000 | 0.000 | -0.000 |
| 10.711 | 10.711 | 76.093 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 10.089 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 10.089 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 10.089 |
| 76.092 | 10.710 | 10.710 | -0.000 | -0.000 | -0.000 |
| 10.710 | 76.092 | 10.710 | -0.000 | -0.000 | 0.000 |
| 10.710 | 10.710 | 76.092 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 10.089 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 10.089 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 10.089 |
| 402.875 | 579.211 | 579.211 | 0.000 | 0.000 | -0.000 |
| 579.211 | 402.875 | 579.211 | -0.000 | 0.000 | -0.000 |
| 579.211 | 579.211 | 402.875 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 323.496 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 323.496 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 323.496 |
| 402.875 | 579.211 | 579.211 | -0.000 | 0.000 | 0.000 |
| 579.211 | 402.875 | 579.211 | 0.000 | 0.000 | 0.000 |
| 579.211 | 579.211 | 402.875 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 323.496 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 323.496 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 323.496 |
| 402.875 | 579.211 | 579.211 | -0.000 | 0.000 | 0.000 |
| 579.211 | 402.875 | 579.211 | -0.000 | -0.000 | 0.000 |
| 579.211 | 579.211 | 402.875 | -0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 323.496 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 323.496 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 323.496 |
| 402.876 | 579.211 | 579.211 | 0.000 | 0.000 | -0.000 |
| 579.211 | 402.876 | 579.211 | 0.000 | 0.000 | 0.000 |
| 579.211 | 579.211 | 402.876 | 0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 323.496 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 323.496 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 323.496 |
| 402.872 | 579.210 | 579.210 | -0.000 | 0.000 | -0.000 |
| 579.210 | 402.872 | 579.210 | 0.000 | 0.000 | 0.000 |
| 579.210 | 579.210 | 402.872 | -0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 323.496 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 323.496 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 323.496 |
| 402.878 | 579.212 | 579.212 | -0.000 | 0.000 | 0.000 |
| 579.212 | 402.878 | 579.212 | 0.000 | 0.000 | 0.000 |
| 579.212 | 579.212 | 402.878 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 323.496 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 323.496 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 323.496 |
| 30.997 | 36.718 | 36.718 | -0.000 | -0.000 | -0.000 |
| 36.718 | 30.997 | 36.718 | -0.000 | -0.000 | 0.000 |
| 36.718 | 36.718 | 30.997 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 31.904 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 31.904 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 31.904 |
| 30.997 | 36.718 | 36.718 | -0.000 | 0.000 | 0.000 |
| 36.718 | 30.997 | 36.718 | 0.000 | 0.000 | 0.000 |
| 36.718 | 36.718 | 30.997 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 31.904 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 31.904 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 31.904 |
| 30.997 | 36.718 | 36.718 | -0.000 | -0.000 | -0.000 |
| 36.718 | 30.997 | 36.718 | -0.000 | -0.000 | -0.000 |
| 36.718 | 36.718 | 30.997 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 31.904 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 31.904 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 31.904 |
| 30.997 | 36.718 | 36.718 | 0.000 | 0.000 | 0.000 |
| 36.718 | 30.997 | 36.718 | 0.000 | 0.000 | -0.000 |
| 36.718 | 36.718 | 30.997 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 31.904 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 31.904 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 31.904 |
| 30.997 | 36.718 | 36.718 | -0.000 | -0.000 | -0.000 |
| 36.718 | 30.997 | 36.718 | -0.000 | -0.000 | -0.000 |
| 36.718 | 36.718 | 30.997 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 31.904 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 31.904 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 31.904 |
| 30.996 | 36.718 | 36.718 | 0.000 | 0.000 | 0.000 |
| 36.718 | 30.996 | 36.718 | 0.000 | 0.000 | 0.000 |
| 36.718 | 36.718 | 30.996 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 31.904 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 31.904 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 31.904 |
| 176.920 | 120.338 | 120.338 | -0.000 | -0.000 | 0.000 |
| 120.338 | 176.920 | 120.338 | -0.000 | -0.000 | -0.000 |
| 120.338 | 120.338 | 176.920 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 86.535 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 86.535 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 86.535 |
| 176.920 | 120.338 | 120.338 | 0.000 | -0.000 | -0.000 |
| 120.338 | 176.920 | 120.338 | -0.000 | 0.000 | -0.000 |
| 120.338 | 120.338 | 176.920 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 86.535 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 86.535 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 86.535 |
| 176.920 | 120.338 | 120.338 | -0.000 | -0.000 | 0.000 |
| 120.338 | 176.920 | 120.338 | -0.000 | -0.000 | -0.000 |
| 120.338 | 120.338 | 176.920 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 75.482 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 75.482 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 86.535 |
| 176.920 | 120.338 | 120.338 | 0.000 | 0.000 | 0.000 |
| 120.338 | 176.920 | 120.338 | 0.000 | 0.000 | 0.000 |
| 120.338 | 120.338 | 176.920 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 86.535 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 86.535 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 86.535 |
| 176.920 | 120.338 | 120.338 | -0.000 | -0.000 | -0.000 |
| 120.338 | 176.920 | 120.338 | -0.000 | -0.000 | -0.000 |
| 120.338 | 120.338 | 176.920 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 75.482 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 75.482 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 75.482 |
| 176.920 | 120.337 | 120.337 | 0.000 | 0.000 | 0.000 |
| 120.337 | 176.920 | 120.337 | 0.000 | 0.000 | 0.000 |
| 120.337 | 120.337 | 176.920 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 75.482 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 75.482 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 75.482 |
| -244.736 | -177.218 | -177.218 | 0.000 | 0.000 | 0.000 |
| -177.218 | -244.736 | -177.218 | 0.000 | 0.000 | 0.000 |
| -177.218 | -177.218 | -244.736 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -90.753 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -90.753 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -90.753 |
| -244.736 | -177.218 | -177.217 | -0.000 | -0.000 | -0.000 |
| -177.217 | -244.736 | -177.217 | -0.000 | -0.000 | -0.000 |
| -177.217 | -177.218 | -244.736 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -90.753 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -90.753 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -90.753 |
| -244.736 | -177.217 | -177.217 | 0.000 | 0.000 | 0.000 |
| -177.217 | -244.736 | -177.217 | 0.000 | 0.000 | 0.000 |
| -177.217 | -177.217 | -244.736 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -90.753 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -90.753 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | -90.753 |
| -244.736 | -177.217 | -177.217 | -0.000 | -0.000 | -0.000 |
| -177.217 | -244.736 | -177.217 | -0.000 | -0.000 | -0.000 |
| -177.217 | -177.217 | -244.736 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -90.753 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -90.753 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -90.753 |
| -244.737 | -177.218 | -177.218 | 0.000 | 0.000 | 0.000 |
| -177.218 | -244.737 | -177.218 | 0.000 | 0.000 | 0.000 |
| -177.218 | -177.218 | -244.737 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -90.753 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -90.753 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | -90.753 |
| -244.736 | -177.217 | -177.217 | 0.000 | 0.000 | 0.000 |
| -177.217 | -244.736 | -177.217 | 0.000 | 0.000 | 0.000 |
| -177.217 | -177.217 | -244.736 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -90.753 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -90.753 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -90.753 |
| -97.234 | -69.737 | -69.737 | 0.000 | 0.000 | 0.000 |
| -69.737 | -97.234 | -69.737 | 0.000 | 0.000 | 0.000 |
| -69.737 | -69.737 | -97.234 | 0.002 | 0.002 | 0.002 |
| 0.000 | -0.000 | -0.000 | -676.760 | 0.002 | 0.002 |
| -0.000 | 0.000 | -0.000 | 0.002 | -676.760 | 0.002 |
| -0.000 | -0.000 | 0.000 | 0.002 | 0.002 | -676.760 |
| -97.234 | -69.737 | -69.737 | -0.002 | -0.002 | -0.002 |
| -69.737 | -97.234 | -69.737 | -0.002 | -0.002 | -0.002 |
| -69.737 | -69.737 | -97.234 | -0.002 | -0.002 | -0.002 |
| -0.000 | 0.000 | 0.000 | -676.764 | -0.002 | -0.002 |
| 0.000 | -0.000 | 0.000 | -0.002 | -676.764 | -0.002 |
| 0.000 | 0.000 | -0.000 | -0.002 | -0.002 | -676.764 |
| -97.234 | -69.737 | -69.737 | 0.000 | 0.000 | 0.000 |
| -69.737 | -97.234 | -69.737 | 0.000 | 0.000 | 0.000 |
| -69.737 | -69.737 | -97.234 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -70.164 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -70.164 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -70.164 |
| -97.234 | -69.737 | -69.737 | -0.000 | -0.000 | -0.000 |
| -69.737 | -97.234 | -69.737 | -0.000 | -0.000 | -0.000 |
| -69.737 | -69.737 | -97.234 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -70.164 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -70.164 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -70.164 |
| -97.234 | -69.737 | -69.737 | 0.000 | 0.000 | 0.000 |
| -69.737 | -97.234 | -69.737 | 0.000 | 0.000 | 0.000 |
| -69.737 | -69.737 | -97.234 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -70.164 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -70.164 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -70.164 |
| -97.234 | -69.737 | -69.737 | -0.000 | -0.000 | -0.000 |
| -69.737 | -97.234 | -69.737 | -0.000 | -0.000 | -0.000 |
| -69.737 | -69.737 | -97.234 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -70.164 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -70.164 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -70.164 |
| 7.834 | 7.834 | 7.834 | -0.000 | -0.000 | 0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | -0.000 | -0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 7.834 | 7.834 | 7.834 | 0.000 | -0.000 | -0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | 0.000 | -0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | -0.000 | 0.000 |
| 7.834 | 7.834 | 7.834 | 0.000 | -0.000 | 0.000 |
| 7.834 | 7.834 | 7.834 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | -0.000 | 0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | -0.000 | 0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 0.000 |
| 7.834 | 7.834 | 7.834 | 0.000 | -0.000 | 0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | 0.000 | -0.000 |
| 7.834 | 7.834 | 7.834 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | -0.000 | -0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | -0.000 | 0.000 |
| 7.834 | 7.834 | 7.834 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 0.000 |
| 1237.312 | 197.227 | 494.433 | 0.000 | 0.000 | 0.000 |
| 197.227 | 1237.312 | 494.433 | 0.000 | 0.000 | -0.000 |
| 494.433 | 494.433 | 1322.223 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 209.336 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 209.336 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -133.730 |
| 1237.311 | 197.227 | 494.433 | -0.000 | -0.000 | -0.000 |
| 197.227 | 1237.311 | 494.433 | -0.000 | -0.000 | -0.000 |
| 494.433 | 494.433 | 1322.223 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 209.336 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 209.336 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -133.730 |
| 1237.313 | 197.228 | 494.434 | 0.000 | 0.000 | -0.000 |
| 197.228 | 1237.313 | 494.434 | 0.000 | 0.000 | 0.000 |
| 494.434 | 494.434 | 1322.225 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 209.336 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 209.336 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -133.730 |
| 1237.311 | 197.227 | 494.432 | -0.000 | -0.000 | -0.000 |
| 197.227 | 1237.311 | 494.432 | -0.000 | -0.000 | -0.000 |
| 494.432 | 494.432 | 1322.221 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 209.336 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 209.336 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -133.730 |
| 1237.321 | 197.233 | 494.439 | 0.000 | 0.000 | -0.000 |
| 197.233 | 1237.321 | 494.439 | 0.000 | 0.000 | -0.000 |
| 494.443 | 494.443 | 1322.245 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.001 | 209.336 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.001 | 0.000 | 209.336 | 0.000 |
| 0.000 | -0.001 | 0.000 | 0.000 | 0.000 | -133.730 |
| 1237.302 | 197.222 | 494.427 | -0.000 | -0.000 | -0.000 |
| 197.222 | 1237.302 | 494.427 | -0.000 | -0.000 | -0.000 |
| 494.424 | 494.424 | 1322.201 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.001 | 209.336 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.001 | -0.000 | 209.336 | -0.000 |
| -0.000 | 0.001 | -0.000 | -0.000 | -0.000 | -133.730 |
| 79.407 | 20.407 | 31.475 | -0.000 | -0.000 | -0.000 |
| 20.407 | 79.407 | 31.475 | -0.000 | -0.000 | -0.000 |
| 31.475 | 31.475 | 39.325 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 31.395 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 31.395 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 20.344 |
| 79.407 | 20.407 | 31.475 | 0.000 | 0.000 | 0.000 |
| 20.407 | 79.407 | 31.475 | 0.000 | 0.000 | 0.000 |
| 31.475 | 31.475 | 39.325 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 31.395 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 31.395 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 20.344 |
| 79.407 | 20.407 | 31.475 | 0.000 | -0.000 | -0.000 |
| 20.407 | 79.407 | 31.475 | -0.000 | -0.000 | -0.000 |
| 31.475 | 31.475 | 39.326 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 31.395 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 31.395 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 20.344 |
| 79.407 | 20.407 | 31.475 | 0.000 | 0.000 | 0.000 |
| 20.407 | 79.407 | 31.475 | 0.000 | 0.000 | -0.000 |
| 31.475 | 31.475 | 39.325 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 31.395 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 31.395 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 20.344 |
| 79.407 | 20.407 | 31.475 | 0.000 | -0.000 | -0.000 |
| 20.407 | 79.407 | 31.475 | -0.000 | -0.000 | -0.000 |
| 31.475 | 31.475 | 39.326 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 31.395 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 31.395 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 20.344 |
| 79.406 | 20.407 | 31.475 | 0.000 | 0.000 | 0.000 |
| 20.407 | 79.406 | 31.475 | 0.000 | 0.000 | 0.000 |
| 31.475 | 31.475 | 39.325 | -0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 31.395 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 31.395 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 20.344 |
| 203.593 | 230.774 | 230.774 | 0.000 | -0.000 | -0.000 |
| 230.774 | 203.593 | 230.774 | -0.000 | -0.000 | 0.000 |
| 230.774 | 230.774 | 203.593 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 28.605 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 28.605 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 28.605 |
| 203.593 | 230.774 | 230.774 | 0.000 | 0.000 | 0.000 |
| 230.774 | 203.593 | 230.774 | 0.000 | 0.000 | 0.000 |
| 230.774 | 230.774 | 203.593 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 28.605 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 28.605 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 28.605 |
| 203.593 | 230.774 | 230.774 | -0.000 | 0.000 | -0.000 |
| 230.774 | 203.593 | 230.774 | 0.000 | -0.000 | -0.000 |
| 230.774 | 230.774 | 203.593 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 28.605 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 28.605 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 28.605 |
| 203.593 | 230.774 | 230.774 | 0.000 | 0.000 | 0.000 |
| 230.774 | 203.593 | 230.774 | 0.000 | 0.000 | 0.000 |
| 230.774 | 230.774 | 203.593 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 28.605 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 28.605 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 28.605 |
| 203.595 | 230.774 | 230.774 | -0.000 | 0.000 | -0.000 |
| 230.774 | 203.595 | 230.774 | 0.000 | -0.000 | -0.000 |
| 230.774 | 230.774 | 203.595 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 110.280 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 110.280 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 110.280 |
| 203.591 | 230.773 | 230.773 | 0.000 | 0.000 | 0.000 |
| 230.773 | 203.591 | 230.773 | 0.000 | 0.000 | 0.000 |
| 230.773 | 230.773 | 203.591 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 110.280 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 110.280 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 110.280 |
| -357.017 | -242.094 | -242.094 | 0.000 | 0.000 | 0.000 |
| -242.094 | -357.017 | -242.094 | 0.000 | 0.000 | 0.000 |
| -242.094 | -242.094 | -357.017 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -189.295 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -189.295 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -189.295 |
| -357.017 | -242.094 | -242.094 | -0.000 | -0.000 | -0.000 |
| -242.094 | -357.017 | -242.094 | -0.000 | -0.000 | -0.000 |
| -242.094 | -242.094 | -357.017 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -189.295 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -189.295 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -189.295 |
| -357.017 | -242.094 | -242.094 | 0.000 | 0.000 | 0.000 |
| -242.094 | -357.017 | -242.094 | 0.000 | 0.000 | 0.000 |
| -242.094 | -242.094 | -357.017 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -189.295 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -189.295 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -189.295 |
| -357.017 | -242.094 | -242.094 | -0.000 | 0.000 | -0.000 |
| -242.094 | -357.017 | -242.094 | -0.000 | 0.000 | -0.000 |
| -242.094 | -242.094 | -357.017 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -189.295 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -189.295 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -189.295 |
| -357.018 | -242.093 | -242.093 | 0.000 | 0.000 | 0.000 |
| -242.093 | -357.018 | -242.093 | 0.000 | -0.000 | 0.000 |
| -242.093 | -242.093 | -357.018 | 0.000 | 0.000 | 0.000 |
| 0.001 | 0.001 | 0.001 | -193.973 | 0.000 | 0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | -193.973 | 0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | -0.000 | -193.973 |
| -357.016 | -242.095 | -242.095 | -0.000 | -0.000 | -0.000 |
| -242.095 | -357.016 | -242.095 | -0.000 | -0.000 | -0.000 |
| -242.095 | -242.095 | -357.016 | -0.000 | 0.000 | -0.000 |
| -0.001 | -0.001 | -0.001 | -193.973 | -0.000 | -0.000 |
| -0.001 | -0.001 | -0.001 | -0.000 | -193.973 | -0.000 |
| -0.001 | -0.001 | -0.001 | -0.000 | -0.000 | -193.973 |
| -244.746 | -237.339 | -237.339 | -0.000 | -0.000 | -0.000 |
| -237.339 | -244.746 | -237.339 | -0.000 | -0.000 | -0.000 |
| -237.339 | -237.339 | -244.746 | -0.000 | -0.000 | -0.000 |
| -0.001 | -0.001 | -0.001 | -1231.682 | -0.000 | -0.000 |
| -0.001 | -0.001 | -0.001 | -0.000 | -1231.682 | -0.000 |
| -0.001 | -0.001 | -0.001 | -0.000 | -0.000 | -1231.682 |
| -244.746 | -237.339 | -237.339 | 0.000 | 0.000 | 0.000 |
| -237.339 | -244.746 | -237.339 | 0.000 | 0.000 | 0.000 |
| -237.339 | -237.339 | -244.746 | 0.000 | 0.000 | -0.000 |
| 0.001 | 0.001 | 0.001 | -1231.682 | 0.000 | -0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | -1231.682 | 0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | 0.000 | -1231.682 |
| -244.746 | -237.339 | -237.339 | 0.000 | -0.000 | -0.000 |
| -237.339 | -244.746 | -237.339 | -0.000 | 0.000 | -0.000 |
| -237.339 | -237.339 | -244.746 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -241.113 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -241.113 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -241.113 |
| -244.745 | -237.339 | -237.339 | -0.000 | -0.000 | -0.000 |
| -237.339 | -244.745 | -237.339 | -0.000 | 0.000 | 0.000 |
| -237.339 | -237.339 | -244.745 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -241.113 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -241.113 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -241.113 |
| -244.748 | -237.340 | -237.340 | 0.000 | -0.000 | 0.000 |
| -237.340 | -244.748 | -237.340 | 0.000 | -0.000 | 0.000 |
| -237.340 | -237.340 | -244.748 | 0.000 | 0.000 | 0.000 |
| -0.001 | -0.000 | -0.001 | -256.590 | 0.000 | -0.000 |
| -0.000 | -0.001 | -0.001 | 0.000 | -256.590 | 0.000 |
| -0.000 | -0.001 | -0.001 | 0.000 | -0.000 | -256.590 |
| -244.743 | -237.338 | -237.338 | 0.000 | 0.000 | -0.000 |
| -237.338 | -244.743 | -237.338 | 0.000 | 0.000 | -0.000 |
| -237.338 | -237.338 | -244.743 | -0.000 | 0.000 | 0.000 |
| 0.001 | 0.000 | 0.001 | -256.590 | 0.000 | 0.000 |
| 0.000 | 0.001 | 0.001 | 0.000 | -256.590 | 0.000 |
| 0.000 | 0.001 | 0.001 | 0.000 | 0.000 | -256.590 |
| -8.279 | 17.671 | 17.671 | -0.000 | -0.000 | 0.000 |
| 17.671 | -8.279 | 17.671 | -0.000 | -0.000 | 0.000 |
| 17.671 | 17.671 | -8.279 | -0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 17.261 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 17.261 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 17.261 |
| -8.279 | 17.671 | 17.671 | 0.000 | -0.000 | -0.000 |
| 17.671 | -8.279 | 17.671 | -0.000 | 0.000 | -0.000 |
| 17.671 | 17.671 | -8.279 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 17.261 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 17.261 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 17.261 |
| -8.279 | 17.671 | 17.671 | -0.000 | -0.000 | 0.000 |
| 17.671 | -8.279 | 17.671 | -0.000 | -0.000 | -0.000 |
| 17.671 | 17.671 | -8.279 | -0.000 | -0.000 | 0.000 |
| -13951248.198 | -12246440.277 | -12497521.079 | -2763886.211 | 158448.678 | 59206.811 |
| -12638069.775 | -14111534.781 | -12868913.391 | 90078.197 | -2631872.897 | 63671.709 |
| -12431258.192 | -12422979.823 | -14256952.724 | 89770.952 | 130870.733 | -2603915.086 |
| -8.279 | 17.671 | 17.671 | 0.000 | 0.000 | -0.000 |
| 17.671 | -8.279 | 17.671 | 0.000 | 0.000 | -0.000 |
| 17.671 | 17.671 | -8.279 | 0.000 | 0.000 | -0.000 |
| 13511772.976 | 12967742.207 | 13094631.624 | -2562960.494 | 263097.151 | -282803.012 |
| 12770848.815 | 13387564.276 | 13017744.370 | 131992.196 | -2561218.467 | -128338.394 |
| 12718780.205 | 13126530.548 | 13576366.134 | 209328.423 | -95008.663 | -2510682.501 |
| -8.279 | 17.671 | 17.671 | -0.000 | -0.000 | 0.000 |
| 17.671 | -8.279 | 17.671 | -0.000 | -0.000 | 0.000 |
| 17.671 | 17.671 | -8.279 | 0.000 | 0.000 | 0.000 |
| -1452561.756 | -1253076.447 | -1302558.952 | -290376.883 | -23185.149 | -33672.387 |
| -1268317.019 | -1443528.114 | -1331607.386 | -11321.991 | -271193.353 | -24694.957 |
| -1311648.409 | -1299870.520 | -1526890.444 | -330.364 | 8481.407 | -279659.602 |
| -8.279 | 17.671 | 17.671 | 0.000 | -0.000 | -0.000 |
| 17.671 | -8.279 | 17.671 | 0.000 | 0.000 | -0.000 |
| 17.671 | 17.671 | -8.279 | -0.000 | -0.000 | -0.000 |
| 1433494.546 | 1242207.232 | 1283474.208 | -288102.747 | -4291.460 | 22817.792 |
| 1223865.498 | 1417769.998 | 1273645.478 | -16839.860 | -296299.901 | 33681.464 |
| 1193996.586 | 1241990.953 | 1389713.515 | -8391.342 | 14006.968 | -276705.191 |
| 2359.172 | 1082.092 | -852.398 | -0.000 | -0.000 | -0.000 |
| 2691.883 | 1224.460 | 661.003 | -0.000 | 0.000 | -0.000 |
| 313.209 | 326.287 | 544.387 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 139.122 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 123.651 | 0.000 |
| 1804.375 | 258.878 | -1359.614 | -0.000 | -0.000 | 189.636 |
| 819.038 | 420.727 | 313.209 | 0.000 | 0.000 | -0.000 |
| -1928.905 | 128.002 | -318.361 | -0.000 | 0.000 | 0.000 |
| -2058.655 | -336.042 | -330.361 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 139.122 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 123.651 | -0.000 |
| -1804.375 | -258.878 | 1359.614 | -0.000 | -0.000 | 189.636 |
| 819.039 | 420.727 | 313.209 | -0.000 | 0.000 | 0.000 |
| 585.305 | 717.848 | 198.385 | -0.000 | -0.000 | -0.000 |
| 296.825 | 199.052 | 77.766 | -0.000 | -0.000 | -0.000 |
| 230.372 | 54.781 | 49.104 | 134.024 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 123.651 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 200.547 |
| 478.471 | 321.083 | 100.732 | -0.000 | -0.000 | 0.000 |
| 420.727 | 746.858 | 326.288 | -0.000 | 0.000 | -0.000 |
| -24.069 | 133.197 | 277.840 | 0.000 | -0.000 | 0.000 |
| -230.372 | -54.781 | -49.104 | 134.024 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 123.651 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 200.547 |
| 690.604 | 374.932 | 171.480 | 0.000 | -0.000 | -0.000 |
| 383.833 | 700.415 | 228.336 | -0.000 | -0.000 | -0.000 |
| 161.977 | 218.284 | 196.229 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 139.122 | -0.000 | -0.000 |
| 16.949 | 3.084 | -6.086 | -0.000 | 64.092 | -0.000 |
| 0.001 | 0.000 | -0.000 | -0.000 | -0.000 | 200.547 |
| 639.258 | 370.062 | 168.790 | -0.000 | -0.000 | 0.000 |
| 343.882 | 674.913 | 205.110 | -0.000 | 0.000 | 0.000 |
| 313.208 | 326.287 | 544.385 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 139.122 | 0.000 | -0.000 |
| -16.949 | -3.084 | 6.086 | 0.000 | 64.092 | 0.000 |
| -0.001 | -0.000 | 0.000 | 0.000 | 0.000 | 200.547 |
| 330.790 | 292.626 | 279.772 | -0.000 | 0.000 | -0.000 |
| 1024.505 | 957.240 | 1842.789 | -0.000 | 0.000 | -0.000 |
| 497.019 | 449.415 | 721.268 | -0.000 | 0.000 | 0.000 |
| 562.142 | 260.529 | 909.490 | 116.408 | 0.000 | 0.000 |
| 560.843 | 259.927 | 907.388 | -0.000 | -0.591 | 0.000 |
| 562.293 | 260.599 | 909.734 | -0.000 | 0.000 | 55.619 |
| -425.466 | -38.711 | -921.173 | -0.000 | -0.000 | -0.000 |
| -532.949 | 409.160 | -1355.089 | 0.000 | -0.000 | 0.000 |
| 264.105 | 330.459 | 532.814 | 0.000 | -0.000 | -0.000 |
| -562.142 | -260.529 | -909.490 | 116.408 | -0.000 | -0.000 |
| -560.843 | -259.927 | -907.388 | 0.000 | -0.591 | -0.000 |
| -562.293 | -260.599 | -909.734 | -0.000 | -0.000 | 55.619 |
| 330.791 | 292.626 | 279.772 | 0.000 | 0.000 | 0.000 |
| 280.476 | 703.478 | 328.026 | 0.000 | 0.000 | 0.000 |
| 160.272 | 264.848 | 183.695 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 118.350 | 0.000 | -0.000 |
| 49.065 | 22.740 | 79.383 | -0.000 | 8.006 | 0.000 |
| 56.814 | 26.331 | 91.919 | 0.000 | 0.000 | 55.553 |
| 330.790 | 292.626 | 279.772 | 0.000 | -0.000 | -0.000 |
| 181.496 | 649.871 | 96.116 | 0.000 | -0.000 | -0.000 |
| 264.105 | 330.459 | 532.814 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 118.350 | -0.000 | 0.000 |
| -49.065 | -22.740 | -79.383 | -0.000 | 8.006 | -0.000 |
| -56.814 | -26.331 | -91.919 | -0.000 | -0.000 | 55.553 |
| 247.737 | 263.481 | 156.422 | 0.000 | 0.000 | 0.000 |
| 262.923 | 681.694 | 265.094 | 0.000 | 0.000 | -0.000 |
| 264.105 | 330.460 | 532.814 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 118.350 | 0.000 | 0.000 |
| 4.174 | 1.935 | 6.753 | 0.000 | 16.976 | 0.000 |
| 6.599 | 3.058 | 10.674 | 0.000 | 0.000 | 54.525 |
| 233.481 | 257.776 | 134.434 | 0.000 | -0.000 | -0.000 |
| 242.918 | 678.450 | 224.682 | 0.000 | -0.000 | -0.000 |
| 124.295 | 273.502 | 172.469 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 118.350 | -0.000 | 0.000 |
| -4.174 | -1.935 | -6.753 | 0.000 | 16.976 | -0.000 |
| -6.599 | -3.058 | -10.674 | 0.000 | -0.000 | 54.525 |
| 493.678 | 286.034 | 286.034 | -0.000 | -0.000 | -0.000 |
| 286.034 | 493.678 | 286.034 | 0.000 | -0.000 | 0.000 |
| 286.034 | 286.034 | 493.678 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 51.503 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 51.503 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 51.503 |
| 494.564 | 285.591 | 285.591 | -0.000 | -0.000 | -0.000 |
| 285.591 | 494.564 | 285.591 | 0.000 | -0.000 | -0.000 |
| 285.591 | 285.591 | 494.564 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 51.503 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 51.503 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 51.503 |
| 493.678 | 286.034 | 286.034 | 0.000 | -0.000 | 0.000 |
| 286.034 | 493.678 | 286.034 | 0.000 | -0.000 | 0.000 |
| 286.034 | 286.034 | 493.678 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -36.010 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -36.010 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | -36.010 |
| 494.564 | 285.591 | 285.591 | -0.000 | -0.000 | -0.000 |
| 285.591 | 494.564 | 285.591 | -0.000 | -0.000 | 0.000 |
| 285.591 | 285.591 | 494.564 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -36.010 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -36.010 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | -36.010 |
| 494.568 | 285.592 | 285.592 | 0.000 | 0.000 | 0.000 |
| 285.592 | 494.568 | 285.592 | 0.000 | 0.000 | 0.000 |
| 285.592 | 285.592 | 494.568 | 0.000 | 0.000 | 0.000 |
| 4325469.968 | 3251116.381 | 3251108.156 | -4112089.043 | -0.000 | -0.000 |
| 3251116.381 | 4325469.968 | 3251108.155 | 0.000 | -4112089.043 | 0.000 |
| 3251116.381 | 3251108.156 | 4325469.968 | -0.002 | -0.002 | -4112089.043 |
| 493.673 | 286.034 | 286.034 | -0.000 | -0.000 | -0.000 |
| 286.034 | 493.673 | 286.034 | -0.000 | 0.000 | -0.000 |
| 286.034 | 286.034 | 493.673 | 0.000 | -0.000 | 0.000 |
| -4325469.968 | -3251116.381 | -3251108.156 | -4112089.043 | 0.000 | -0.000 |
| -3251116.381 | -4325469.968 | -3251108.155 | -0.001 | -4112089.043 | 0.001 |
| -3251116.381 | -3251108.156 | -4325469.968 | 0.003 | -0.003 | -4112089.043 |
| 49.104 | 15.710 | 15.710 | -0.000 | -0.000 | 0.000 |
| 15.710 | 49.104 | 15.710 | -0.000 | -0.000 | 0.000 |
| 15.710 | 15.710 | 49.104 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -12.851 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -12.851 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -12.851 |
| 49.104 | 15.710 | 15.710 | -0.000 | 0.000 | -0.000 |
| 15.710 | 49.104 | 15.710 | 0.000 | 0.000 | -0.000 |
| 15.710 | 15.710 | 49.104 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -12.851 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -12.851 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -12.851 |
| 58.925 | 10.800 | 10.800 | -0.000 | -0.000 | -0.000 |
| 10.800 | 58.925 | 10.800 | -0.000 | -0.000 | 0.000 |
| 10.800 | 10.800 | 58.925 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 10.400 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 10.400 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -12.851 |
| 49.104 | 15.710 | 15.710 | -0.000 | 0.000 | -0.000 |
| 15.710 | 49.104 | 15.710 | 0.000 | 0.000 | -0.000 |
| 15.710 | 15.710 | 49.104 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 10.400 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 10.400 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -12.851 |
| 58.925 | 10.800 | 10.800 | -0.000 | -0.000 | 0.000 |
| 10.800 | 58.925 | 10.800 | -0.000 | -0.000 | 0.000 |
| 10.800 | 10.800 | 58.925 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -12.851 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -12.851 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -12.851 |
| 49.104 | 15.710 | 15.710 | -0.000 | 0.000 | -0.000 |
| 15.710 | 49.104 | 15.710 | -0.000 | 0.000 | -0.000 |
| 15.710 | 15.710 | 49.104 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -12.851 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -12.851 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -12.851 |
| 559.237 | 556.865 | 556.865 | 0.000 | 0.000 | -0.000 |
| 556.865 | 559.237 | 556.865 | 0.000 | -0.000 | -0.000 |
| 556.865 | 556.865 | 559.237 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 244.375 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 244.375 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 244.375 |
| 559.237 | 556.865 | 556.865 | -0.000 | -0.000 | 0.000 |
| 556.865 | 559.237 | 556.865 | -0.000 | -0.000 | 0.000 |
| 556.865 | 556.865 | 559.237 | -0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 244.375 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 244.375 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 244.375 |
| 559.236 | 556.864 | 556.864 | 0.000 | 0.000 | 0.000 |
| 556.864 | 559.236 | 556.864 | 0.000 | 0.000 | -0.000 |
| 556.864 | 556.864 | 559.236 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 234.743 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 234.743 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 234.743 |
| 559.238 | 556.866 | 556.866 | -0.000 | -0.000 | 0.000 |
| 556.866 | 559.238 | 556.866 | -0.000 | -0.000 | 0.000 |
| 556.866 | 556.866 | 559.238 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 234.743 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 234.743 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 234.743 |
| 559.228 | 556.855 | 556.855 | -0.000 | 0.000 | 0.000 |
| 556.855 | 559.228 | 556.855 | 0.000 | -0.000 | 0.000 |
| 556.855 | 556.855 | 559.228 | 0.000 | 0.000 | -0.000 |
| 0.001 | 0.002 | 0.002 | 244.375 | -0.000 | -0.000 |
| 0.002 | 0.001 | 0.002 | -0.000 | 244.375 | -0.000 |
| 0.002 | 0.002 | 0.001 | -0.000 | -0.000 | 244.375 |
| 559.246 | 556.876 | 556.876 | -0.000 | 0.000 | 0.000 |
| 556.876 | 559.246 | 556.876 | -0.000 | -0.000 | 0.000 |
| 556.876 | 556.876 | 559.246 | 0.000 | 0.000 | 0.000 |
| -0.001 | -0.002 | -0.002 | 244.375 | -0.000 | -0.000 |
| -0.002 | -0.001 | -0.002 | -0.000 | 244.375 | -0.000 |
| -0.002 | -0.002 | -0.001 | 0.000 | 0.000 | 244.375 |
| 20.646 | -308.847 | -308.847 | -0.000 | 0.000 | 0.000 |
| -308.847 | 20.646 | -308.847 | 0.000 | 0.000 | -0.000 |
| -308.847 | -308.847 | 20.646 | -0.000 | -0.000 | -0.000 |
| 0.068 | 0.068 | 0.068 | -582.220 | -0.000 | -0.000 |
| 0.068 | 0.068 | 0.068 | -0.000 | -582.220 | -0.000 |
| 0.068 | 0.068 | 0.068 | -0.000 | -0.000 | -582.220 |
| 20.626 | -308.866 | -308.866 | 0.000 | 0.000 | -0.000 |
| -308.866 | 20.626 | -308.866 | 0.000 | 0.000 | 0.000 |
| -308.866 | -308.866 | 20.626 | 0.000 | 0.000 | 0.000 |
| -0.068 | -0.068 | -0.068 | -582.220 | 0.000 | 0.000 |
| -0.068 | -0.068 | -0.068 | 0.000 | -582.220 | 0.000 |
| -0.068 | -0.068 | -0.068 | 0.000 | 0.000 | -582.220 |
| 20.646 | -308.847 | -308.847 | 0.000 | 0.000 | -0.000 |
| -308.847 | 20.646 | -308.847 | -0.000 | 0.000 | -0.000 |
| -308.847 | -308.847 | 20.646 | 0.000 | 0.000 | -0.000 |
| 0.010 | 0.010 | 0.010 | -279.820 | -0.000 | -0.000 |
| 0.010 | 0.010 | 0.010 | -0.000 | -279.820 | -0.000 |
| 0.010 | 0.010 | 0.010 | -0.000 | -0.000 | -279.820 |
| 20.626 | -308.866 | -308.866 | -0.000 | -0.000 | 0.000 |
| -308.866 | 20.626 | -308.866 | -0.000 | -0.000 | 0.000 |
| -308.866 | -308.866 | 20.626 | 0.000 | 0.000 | 0.000 |
| -0.010 | -0.010 | -0.010 | -279.820 | 0.000 | 0.000 |
| -0.010 | -0.010 | -0.010 | 0.000 | -279.820 | 0.000 |
| -0.010 | -0.010 | -0.010 | 0.000 | 0.000 | -279.820 |
| 20.646 | -308.847 | -308.847 | -0.000 | -0.000 | 0.000 |
| -308.847 | 20.646 | -308.847 | -0.000 | -0.000 | -0.000 |
| -308.847 | -308.847 | 20.646 | -0.000 | -0.000 | -0.000 |
| 0.010 | 0.010 | 0.010 | -279.820 | -0.000 | -0.000 |
| 0.010 | 0.010 | 0.010 | -0.000 | -279.820 | -0.000 |
| 0.010 | 0.010 | 0.010 | -0.000 | -0.000 | -279.820 |
| 20.626 | -308.866 | -308.866 | 0.000 | 0.000 | 0.000 |
| -308.866 | 20.626 | -308.866 | 0.000 | 0.000 | 0.000 |
| -308.866 | -308.866 | 20.626 | 0.000 | 0.000 | 0.000 |
| -0.010 | -0.010 | -0.010 | -279.820 | 0.000 | 0.000 |
| -0.010 | -0.010 | -0.010 | 0.000 | -279.820 | 0.000 |
| -0.010 | -0.010 | -0.010 | 0.000 | 0.000 | -279.820 |
| -389.580 | -303.292 | -303.292 | -0.000 | -0.000 | -0.000 |
| -303.292 | -389.580 | -303.292 | -0.000 | 0.000 | -0.000 |
| -303.292 | -303.292 | -389.580 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -305.718 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -305.718 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | -305.718 |
| -389.580 | -303.292 | -303.292 | 0.000 | 0.000 | 0.000 |
| -303.292 | -389.580 | -303.292 | 0.000 | -0.000 | 0.000 |
| -303.292 | -303.292 | -389.580 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -305.718 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -305.718 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | -305.718 |
| -389.578 | -303.292 | -303.292 | -0.000 | 0.000 | -0.000 |
| -303.292 | -389.578 | -303.292 | 0.000 | 0.000 | 0.000 |
| -303.292 | -303.292 | -389.578 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -305.718 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -305.718 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | -305.718 |
| -389.581 | -303.292 | -303.292 | -0.000 | -0.000 | -0.000 |
| -303.292 | -389.581 | -303.292 | -0.000 | -0.000 | 0.000 |
| -303.292 | -303.292 | -389.581 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -305.718 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -305.718 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | -305.718 |
| -389.566 | -303.291 | -303.291 | 0.000 | 0.000 | -0.000 |
| -303.291 | -389.566 | -303.291 | -0.000 | 0.000 | -0.000 |
| -303.291 | -303.291 | -389.566 | 0.000 | 0.000 | 0.000 |
| 0.001 | 0.002 | 0.002 | -317.564 | 0.000 | -0.000 |
| 0.002 | 0.001 | 0.002 | -0.000 | -317.564 | -0.000 |
| 0.002 | 0.002 | 0.001 | -0.000 | 0.000 | -317.564 |
| -389.593 | -303.293 | -303.293 | -0.000 | -0.000 | 0.000 |
| -303.293 | -389.593 | -303.293 | -0.000 | -0.000 | -0.000 |
| -303.293 | -303.293 | -389.593 | -0.000 | -0.000 | -0.000 |
| -0.001 | -0.002 | -0.002 | -317.564 | -0.000 | 0.000 |
| -0.002 | -0.001 | -0.002 | 0.000 | -317.564 | -0.000 |
| -0.002 | -0.002 | -0.001 | -0.000 | 0.000 | -317.564 |
| 33.465 | 31.223 | 31.223 | -0.000 | 0.000 | -0.000 |
| 31.223 | 33.465 | 31.223 | -0.000 | -0.000 | -0.000 |
| 31.223 | 31.223 | 33.465 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 29.529 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 29.529 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 29.529 |
| 33.465 | 31.223 | 31.223 | 0.000 | -0.000 | 0.000 |
| 31.223 | 33.465 | 31.223 | 0.000 | -0.000 | 0.000 |
| 31.223 | 31.223 | 33.465 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 29.529 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 29.529 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 29.529 |
| 33.465 | 31.223 | 31.223 | -0.000 | -0.000 | -0.000 |
| 31.223 | 33.465 | 31.223 | -0.000 | 0.000 | -0.000 |
| 31.223 | 31.223 | 33.465 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 28.400 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 28.400 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 28.400 |
| 33.465 | 31.223 | 31.223 | 0.000 | 0.000 | 0.000 |
| 31.223 | 33.465 | 31.223 | 0.000 | 0.000 | 0.000 |
| 31.223 | 31.223 | 33.465 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 28.400 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 28.400 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 28.400 |
| 33.466 | 31.223 | 31.223 | -0.000 | -0.000 | -0.000 |
| 31.223 | 33.466 | 31.223 | -0.000 | 0.000 | 0.000 |
| 31.223 | 31.223 | 33.466 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 28.400 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 28.400 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 28.400 |
| 33.465 | 31.223 | 31.223 | 0.000 | -0.000 | -0.000 |
| 31.223 | 33.465 | 31.223 | -0.000 | -0.000 | -0.000 |
| 31.223 | 31.223 | 33.465 | 0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 28.400 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 28.400 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 28.400 |
| 459.935 | 250.479 | 250.479 | -0.000 | -0.000 | -0.000 |
| 250.479 | 459.935 | 250.479 | -0.000 | -0.000 | -0.000 |
| 250.479 | 250.479 | 459.935 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 86.731 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 86.731 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 86.731 |
| 459.935 | 250.479 | 250.479 | 0.000 | 0.000 | 0.000 |
| 250.479 | 459.935 | 250.479 | 0.000 | 0.000 | 0.000 |
| 250.479 | 250.479 | 459.935 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 86.731 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 86.731 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 86.731 |
| 459.935 | 250.479 | 250.479 | -0.000 | -0.000 | -0.000 |
| 250.479 | 459.935 | 250.479 | -0.000 | -0.000 | -0.000 |
| 250.479 | 250.479 | 459.935 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 86.731 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 86.731 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 86.731 |
| 459.935 | 250.479 | 250.479 | 0.000 | 0.000 | 0.000 |
| 250.479 | 459.935 | 250.479 | 0.000 | 0.000 | 0.000 |
| 250.479 | 250.479 | 459.935 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 86.731 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 86.731 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 86.731 |
| 459.933 | 250.480 | 250.480 | -0.000 | -0.000 | -0.000 |
| 250.480 | 459.933 | 250.480 | -0.000 | -0.000 | -0.000 |
| 250.480 | 250.480 | 459.933 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 86.731 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 86.731 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 86.731 |
| 459.937 | 250.479 | 250.479 | 0.000 | 0.000 | 0.000 |
| 250.479 | 459.937 | 250.479 | 0.000 | 0.000 | 0.000 |
| 250.479 | 250.479 | 459.937 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 86.731 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 86.731 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 86.731 |
| 14.835 | 101.299 | 101.299 | 0.000 | 0.000 | 0.000 |
| 101.299 | 14.835 | 101.299 | 0.000 | 0.000 | 0.000 |
| 101.299 | 101.299 | 14.835 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 70.184 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 70.184 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 70.184 |
| 14.835 | 101.299 | 101.299 | 0.000 | -0.000 | -0.000 |
| 101.299 | 14.835 | 101.299 | -0.000 | 0.000 | -0.000 |
| 101.299 | 101.299 | 14.835 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 70.184 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 70.184 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 70.184 |
| 14.835 | 101.299 | 101.299 | 0.000 | 0.000 | 0.000 |
| 101.299 | 14.835 | 101.299 | 0.000 | 0.000 | 0.000 |
| 101.299 | 101.299 | 14.835 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 70.184 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 70.184 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 70.184 |
| 14.834 | 101.299 | 101.299 | -0.000 | -0.000 | -0.000 |
| 101.299 | 14.834 | 101.299 | -0.000 | -0.000 | -0.000 |
| 101.299 | 101.299 | 14.834 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 70.184 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 70.184 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 70.184 |
| 14.836 | 101.299 | 101.299 | 0.000 | 0.000 | 0.000 |
| 101.299 | 14.836 | 101.299 | 0.000 | 0.000 | 0.000 |
| 101.299 | 101.299 | 14.836 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 70.184 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 70.184 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 70.184 |
| 14.833 | 101.299 | 101.299 | -0.000 | -0.000 | -0.000 |
| 101.299 | 14.833 | 101.299 | -0.000 | -0.000 | -0.000 |
| 101.299 | 101.299 | 14.833 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 70.184 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 70.184 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 70.184 |
| 116.294 | 29.800 | 29.800 | 0.000 | 0.000 | 0.000 |
| 29.800 | 116.294 | 29.800 | 0.000 | -0.000 | 0.000 |
| 29.800 | 29.800 | 116.294 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 30.345 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 30.345 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 30.345 |
| 116.294 | 29.800 | 29.800 | 0.000 | 0.000 | 0.000 |
| 29.800 | 116.294 | 29.800 | 0.000 | 0.000 | -0.000 |
| 29.800 | 29.800 | 116.294 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 30.345 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 30.345 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 30.345 |
| 116.294 | 29.800 | 29.800 | -0.000 | -0.000 | 0.000 |
| 29.800 | 116.294 | 29.800 | -0.000 | -0.000 | 0.000 |
| 29.800 | 29.800 | 116.294 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 30.345 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 30.345 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 30.345 |
| 116.294 | 29.800 | 29.800 | 0.000 | -0.000 | -0.000 |
| 29.800 | 116.294 | 29.800 | -0.000 | 0.000 | 0.000 |
| 29.800 | 29.800 | 116.294 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 30.345 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 30.345 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 30.345 |
| 116.294 | 29.800 | 29.800 | 0.000 | 0.000 | 0.000 |
| 29.800 | 116.294 | 29.800 | 0.000 | -0.000 | -0.000 |
| 29.800 | 29.800 | 116.294 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 30.345 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 30.345 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 30.345 |
| 116.294 | 29.800 | 29.800 | -0.000 | 0.000 | -0.000 |
| 29.800 | 116.294 | 29.800 | 0.000 | -0.000 | -0.000 |
| 29.800 | 29.800 | 116.294 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 30.345 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 30.345 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 30.345 |
| 564.493 | 313.462 | 396.427 | -0.000 | -0.000 | 0.000 |
| 314.950 | 553.197 | 397.452 | 0.000 | -0.000 | 0.000 |
| 396.987 | 396.987 | 636.942 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 177.291 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 177.291 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 111.485 |
| 564.493 | 313.462 | 396.427 | 0.000 | 0.000 | -0.000 |
| 313.462 | 564.493 | 396.427 | 0.000 | -0.000 | -0.000 |
| 396.987 | 396.987 | 636.942 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 177.291 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 177.291 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 111.485 |
| 553.197 | 314.950 | 397.452 | 0.000 | -0.000 | 0.000 |
| 313.462 | 564.493 | 396.427 | 0.000 | 0.000 | 0.000 |
| 397.586 | 397.586 | 636.816 | -0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 170.736 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 170.736 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 125.516 |
| 564.493 | 313.462 | 396.428 | -0.000 | -0.000 | -0.000 |
| 313.462 | 564.493 | 396.428 | -0.000 | 0.000 | -0.000 |
| 396.987 | 396.987 | 636.942 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 170.736 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 170.736 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 125.516 |
| 564.491 | 313.459 | 396.424 | 0.000 | 0.000 | 0.000 |
| 313.460 | 564.490 | 396.424 | 0.000 | 0.000 | 0.000 |
| 397.584 | 397.584 | 636.815 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.001 | 177.291 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.001 | -0.000 | 170.736 | 0.000 |
| -0.001 | -0.000 | -0.000 | -0.000 | 0.000 | 111.485 |
| 553.200 | 314.952 | 397.455 | 0.000 | 0.000 | -0.000 |
| 325.349 | 546.980 | 397.019 | -0.000 | -0.000 | -0.000 |
| 396.990 | 396.990 | 636.943 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.001 | 177.291 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.001 | -0.000 | 170.736 | -0.000 |
| 0.001 | 0.000 | 0.000 | 0.000 | -0.000 | 111.485 |
| 547.684 | 326.346 | 400.787 | -0.000 | -0.000 | 0.000 |
| 313.694 | 564.368 | 400.369 | -0.000 | 0.000 | 0.000 |
| 400.369 | 400.369 | 649.725 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 179.403 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 179.403 | 0.000 |
| 2.042 | 2.042 | -0.423 | 0.000 | 0.000 | 112.419 |
| 564.368 | 313.695 | 400.369 | 0.000 | -0.000 | -0.000 |
| 325.132 | 546.470 | 401.038 | 0.000 | -0.000 | -0.000 |
| 400.369 | 400.369 | 649.725 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 179.403 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 179.403 | 0.000 |
| -2.042 | -2.042 | 0.423 | -0.000 | -0.000 | 112.419 |
| 564.368 | 313.694 | 400.368 | 0.000 | 0.000 | 0.000 |
| 325.797 | 547.136 | 400.900 | -0.000 | -0.000 | 0.000 |
| 400.982 | 400.982 | 649.598 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 179.403 | -0.000 | 0.000 |
| 0.220 | 0.220 | -0.046 | 0.000 | 172.977 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 125.337 |
| 553.125 | 315.505 | 401.347 | 0.000 | -0.000 | -0.000 |
| 313.695 | 564.368 | 400.369 | -0.000 | -0.000 | -0.000 |
| 400.863 | 400.863 | 649.622 | 0.000 | -0.000 | -0.000 |
| -0.220 | -0.220 | 0.046 | 172.977 | -0.000 | -0.000 |
| -0.220 | -0.220 | 0.046 | 0.000 | 172.977 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 125.337 |
| 564.366 | 313.692 | 400.366 | 0.000 | 0.000 | 0.000 |
| 325.741 | 547.078 | 400.908 | 0.000 | -0.000 | 0.000 |
| 400.366 | 400.366 | 649.725 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 179.403 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 179.403 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 125.337 |
| 546.980 | 325.737 | 400.927 | 0.000 | -0.000 | -0.000 |
| 313.696 | 564.370 | 400.372 | -0.000 | -0.000 | -0.000 |
| 400.919 | 400.919 | 649.610 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 179.403 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 179.403 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 125.337 |
| 6087.489 | 5881.498 | -9553.130 | 35.595 | -0.000 | -0.000 |
| 5880.917 | 6087.135 | -9552.301 | -35.582 | 0.000 | -0.000 |
| 5818.784 | 5818.784 | -9346.639 | -0.000 | -0.000 | -0.000 |
| 32.293 | -32.293 | 0.000 | 97.168 | 0.000 | -0.000 |
| 5542.751 | 5542.751 | -9826.538 | -0.000 | 97.049 | 33.283 |
| 5543.047 | 5543.047 | -9827.062 | -0.000 | 35.595 | 102.996 |
| -4979.889 | -5224.329 | 10101.896 | 33.283 | 0.000 | 0.000 |
| -5205.543 | -4999.325 | 10102.472 | -35.582 | -0.000 | 0.000 |
| -5267.666 | -5267.666 | 10308.117 | 0.000 | -0.000 | -0.000 |
| 32.293 | -32.293 | -0.000 | 97.168 | 0.000 | 0.000 |
| -5542.751 | -5542.751 | 9826.538 | 0.000 | 97.049 | 33.283 |
| -5543.047 | -5543.047 | 9827.062 | 0.000 | 35.595 | 102.996 |
| 1117.313 | 872.873 | -707.606 | 33.283 | -0.000 | 0.000 |
| 872.873 | 1117.313 | -707.606 | -33.283 | 0.000 | -0.000 |
| 829.360 | 829.360 | -501.075 | -0.000 | -0.000 | 0.000 |
| 587.559 | 520.992 | -982.654 | 97.049 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 97.168 | 32.293 |
| 554.275 | 554.275 | -982.654 | 0.000 | 35.595 | 102.997 |
| 573.281 | 312.367 | 271.488 | 32.293 | 0.000 | -0.000 |
| -235.720 | 8.720 | 1257.776 | -33.283 | 0.000 | 0.000 |
| 271.488 | 271.488 | 487.953 | 0.000 | 0.000 | -0.000 |
| -520.992 | -587.559 | 982.654 | 97.049 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 97.168 | 32.293 |
| -554.275 | -554.275 | 982.654 | -0.000 | 35.595 | 102.997 |
| 599.225 | 393.231 | 176.817 | 35.596 | 0.000 | -0.000 |
| 374.257 | 618.633 | 176.432 | -33.289 | 0.000 | -0.000 |
| 330.513 | 330.513 | 383.313 | -0.000 | -0.000 | 0.000 |
| 91.022 | 19.832 | -98.266 | 96.770 | 0.000 | -0.000 |
| 55.430 | 55.430 | -98.271 | 0.000 | 97.049 | 33.283 |
| 55.639 | 55.639 | -98.639 | 0.000 | 33.287 | 122.189 |
| 488.361 | 282.371 | 373.350 | 35.594 | -0.000 | 0.000 |
| 282.371 | 488.361 | 373.350 | -35.593 | -0.000 | 0.000 |
| 271.488 | 271.488 | 487.954 | -0.000 | 0.000 | 0.000 |
| -19.842 | -91.032 | 98.282 | 96.771 | -0.000 | 0.000 |
| -55.430 | -55.430 | 98.271 | -0.000 | 97.049 | 33.283 |
| -55.639 | -55.639 | 98.639 | -0.000 | 33.287 | 122.189 |
| 513.514 | 324.002 | 289.716 | 31.672 | -0.000 | 0.000 |
| 304.027 | 554.867 | 271.780 | -29.225 | -0.000 | 0.000 |
| 269.611 | 269.611 | 482.120 | 0.000 | 0.000 | 0.000 |
| 21.601 | -38.287 | 13.999 | 96.061 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 96.118 | 29.224 |
| -8.331 | -8.331 | 13.980 | 0.000 | 31.672 | 94.756 |
| 554.867 | 304.027 | 271.780 | 29.224 | -0.000 | 0.000 |
| 340.665 | 530.177 | 261.756 | -31.672 | 0.000 | -0.000 |
| 281.898 | 281.898 | 461.503 | -0.000 | 0.000 | -0.000 |
| 38.287 | -21.601 | -13.999 | 96.061 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 96.118 | 29.224 |
| 8.331 | 8.331 | -13.980 | -0.000 | 31.672 | 94.756 |
| 554.867 | 304.027 | 271.780 | 29.225 | 0.000 | -0.000 |
| 304.027 | 554.867 | 271.780 | -29.225 | -0.000 | 0.000 |
| 272.733 | 272.733 | 476.881 | 0.000 | 0.000 | 0.000 |
| 29.095 | -30.978 | 1.580 | 96.053 | 0.000 | 0.000 |
| -0.942 | -0.942 | 1.580 | 0.000 | 96.053 | 30.037 |
| -0.833 | -0.833 | 1.398 | 0.000 | 31.672 | 94.756 |
| 525.889 | 324.613 | 278.820 | 31.203 | 0.000 | 0.000 |
| 304.027 | 554.866 | 271.780 | -29.224 | 0.000 | -0.000 |
| 269.611 | 269.611 | 482.120 | 0.000 | -0.000 | 0.000 |
| 30.978 | -29.095 | -1.580 | 96.053 | 0.000 | -0.000 |
| 0.942 | 0.942 | -1.580 | -0.000 | 96.053 | 30.037 |
| 0.833 | 0.833 | -1.398 | -0.000 | 31.672 | 94.756 |
| 554.868 | 304.027 | 271.780 | 29.225 | -0.000 | -0.000 |
| 304.027 | 554.868 | 271.780 | -29.226 | -0.000 | 0.000 |
| 269.611 | 269.611 | 482.121 | 0.000 | 0.000 | 0.000 |
| 29.224 | -29.225 | -0.000 | 96.118 | -0.000 | 0.000 |
| -0.254 | -0.254 | 0.426 | 0.000 | 95.943 | 31.415 |
| -0.000 | 0.000 | -0.000 | 0.000 | 29.224 | 125.420 |
| 528.760 | 319.604 | 280.613 | 30.887 | -0.000 | -0.000 |
| 304.026 | 554.865 | 271.780 | -29.223 | 0.000 | -0.000 |
| 269.610 | 269.610 | 482.119 | -0.000 | -0.000 | -0.000 |
| 31.669 | -31.161 | -0.426 | 95.944 | 0.000 | -0.000 |
| 0.254 | 0.254 | -0.426 | -0.000 | 95.943 | 31.415 |
| 0.000 | -0.000 | 0.000 | -0.000 | 29.224 | 125.420 |
| 464.582 | 275.874 | 274.504 | -0.000 | 0.000 | 0.000 |
| 275.874 | 464.582 | 274.504 | 0.000 | -0.000 | -0.000 |
| 275.134 | 275.134 | 550.862 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 104.516 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 104.516 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 100.748 |
| 448.207 | 285.007 | 269.881 | -0.000 | 0.000 | 0.000 |
| 285.007 | 448.207 | 269.881 | 0.000 | -0.000 | -0.000 |
| 273.540 | 273.540 | 541.027 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 104.516 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 104.516 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 100.748 |
| 441.359 | 297.062 | 276.009 | 0.000 | -0.000 | -0.000 |
| 297.062 | 441.359 | 276.009 | 0.000 | 0.000 | 0.000 |
| 275.540 | 275.540 | 543.810 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 104.516 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 104.516 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 100.748 |
| 464.582 | 275.874 | 274.504 | -0.000 | 0.000 | -0.000 |
| 275.874 | 464.582 | 274.504 | 0.000 | -0.000 | -0.000 |
| 275.134 | 275.134 | 550.862 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 104.516 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 104.516 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 100.748 |
| 441.201 | 296.895 | 275.699 | -0.000 | -0.000 | -0.000 |
| 296.895 | 441.201 | 275.699 | -0.000 | 0.000 | -0.000 |
| 275.135 | 275.135 | 550.859 | 0.000 | 0.000 | -0.000 |
| 0.017 | 0.017 | 0.038 | 96.433 | -0.000 | 0.000 |
| 0.017 | 0.017 | 0.038 | 0.000 | 96.433 | 0.000 |
| 0.016 | 0.016 | 0.034 | 0.000 | 0.000 | 100.748 |
| 464.584 | 275.874 | 274.504 | -0.000 | 0.000 | -0.000 |
| 275.874 | 464.584 | 274.504 | 0.000 | -0.000 | -0.000 |
| 275.134 | 275.134 | 550.865 | -0.000 | 0.000 | -0.000 |
| -0.017 | -0.017 | -0.038 | 96.433 | 0.000 | -0.000 |
| -0.017 | -0.017 | -0.038 | -0.000 | 96.433 | 0.000 |
| -0.016 | -0.016 | -0.034 | -0.000 | -0.000 | 100.748 |
| 306.983 | 139.881 | 770.421 | 0.000 | -0.000 | 0.000 |
| 139.881 | 306.983 | 770.421 | 0.000 | 0.000 | -0.000 |
| 275.476 | 275.476 | 537.394 | 0.000 | -0.000 | 0.000 |
| -147.286 | -147.286 | 494.322 | 91.822 | -0.000 | 0.000 |
| -147.286 | -147.286 | 494.322 | 0.000 | 91.822 | -0.000 |
| -147.314 | -147.314 | 494.416 | -0.000 | 0.000 | 99.814 |
| 601.658 | 434.536 | -218.531 | 0.000 | 0.000 | -0.000 |
| 434.536 | 601.658 | -218.531 | -0.000 | 0.000 | 0.000 |
| 275.476 | 275.476 | 537.394 | 0.000 | 0.000 | 0.000 |
| 147.286 | 147.286 | -494.322 | 91.822 | -0.000 | -0.000 |
| 147.286 | 147.286 | -494.322 | -0.000 | 91.822 | -0.000 |
| 147.314 | 147.314 | -494.416 | -0.000 | 0.000 | 99.814 |
| 469.284 | 275.252 | 275.476 | -0.000 | -0.000 | 0.000 |
| 275.252 | 469.284 | 275.476 | -0.000 | -0.000 | -0.000 |
| 275.476 | 275.476 | 537.394 | -0.000 | -0.000 | 0.000 |
| -14.463 | -14.463 | 48.541 | 92.053 | -0.000 | 0.000 |
| -14.463 | -14.463 | 48.541 | 0.000 | 92.053 | -0.000 |
| -14.731 | -14.731 | 49.442 | 0.000 | -0.000 | 99.814 |
| 469.106 | 302.233 | 225.880 | -0.000 | -0.000 | 0.000 |
| 302.233 | 469.106 | 225.880 | -0.000 | -0.000 | -0.000 |
| 290.377 | 290.377 | 479.864 | -0.000 | 0.000 | -0.000 |
| 14.463 | 14.463 | -48.541 | 92.053 | 0.000 | -0.000 |
| 14.463 | 14.463 | -48.541 | 0.000 | 92.053 | 0.000 |
| 14.731 | 14.731 | -49.442 | -0.000 | -0.000 | 99.814 |
| 447.908 | 289.000 | 282.302 | 0.000 | 0.000 | -0.000 |
| 289.000 | 447.908 | 282.302 | 0.000 | 0.000 | -0.000 |
| 274.772 | 274.772 | 534.743 | 0.000 | 0.000 | -0.000 |
| -1.010 | -1.009 | 3.388 | 95.855 | -0.000 | 0.000 |
| -1.009 | -1.010 | 3.388 | 0.000 | 95.855 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 99.814 |
| 450.070 | 294.536 | 268.814 | 0.000 | -0.000 | -0.000 |
| 294.536 | 450.070 | 268.814 | -0.000 | 0.000 | -0.000 |
| 277.105 | 277.105 | 524.909 | 0.000 | -0.000 | 0.000 |
| 1.010 | 1.009 | -3.388 | 95.855 | -0.000 | 0.000 |
| 1.009 | 1.010 | -3.388 | -0.000 | 95.855 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 99.814 |
| 285.698 | 116.113 | 185.271 | -0.000 | 0.000 | 0.000 |
| 116.113 | 285.698 | 185.271 | 0.000 | 0.000 | 0.000 |
| 275.100 | 275.100 | 551.493 | -0.000 | -0.000 | 0.000 |
| -180.470 | -180.470 | -15.883 | 96.856 | 0.000 | 0.000 |
| -180.470 | -180.470 | -15.883 | 0.000 | 96.856 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 100.774 |
| 622.104 | 452.504 | 363.853 | 0.000 | 0.000 | -0.000 |
| 452.504 | 622.104 | 363.853 | 0.000 | 0.000 | -0.000 |
| 275.100 | 275.100 | 551.493 | 0.000 | -0.000 | -0.000 |
| 180.470 | 180.470 | 15.883 | 96.856 | -0.000 | -0.000 |
| 180.470 | 180.470 | 15.883 | 0.000 | 96.856 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 100.774 |
| 464.442 | 275.860 | 274.446 | 0.000 | 0.000 | 0.000 |
| 275.860 | 464.442 | 274.446 | 0.000 | 0.000 | 0.000 |
| 257.791 | 257.791 | 542.369 | -0.000 | 0.000 | 0.000 |
| -16.755 | -16.755 | -8.895 | 95.529 | -0.000 | 0.000 |
| -16.755 | -16.755 | -8.895 | -0.000 | 95.529 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 100.774 |
| 464.442 | 275.859 | 274.446 | 0.000 | 0.000 | -0.000 |
| 275.859 | 464.442 | 274.446 | 0.000 | -0.000 | -0.000 |
| 292.797 | 292.797 | 554.525 | -0.000 | -0.000 | -0.000 |
| 16.755 | 16.755 | 8.895 | 95.529 | -0.000 | -0.000 |
| 16.755 | 16.755 | 8.895 | -0.000 | 95.529 | -0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 100.774 |
| 464.439 | 275.860 | 274.446 | 0.000 | 0.000 | 0.000 |
| 275.860 | 464.439 | 274.446 | -0.000 | 0.000 | -0.000 |
| 275.101 | 275.101 | 551.491 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 104.515 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 104.515 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 100.774 |
| 445.765 | 297.528 | 276.664 | 0.000 | -0.000 | 0.000 |
| 297.528 | 445.765 | 276.664 | -0.000 | 0.000 | -0.000 |
| 277.694 | 277.694 | 544.238 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 104.515 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 104.515 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 100.774 |
| 469.320 | 275.244 | 275.481 | -0.000 | -0.000 | 0.000 |
| 275.244 | 469.320 | 275.481 | -0.000 | -0.000 | 0.000 |
| 275.477 | 275.477 | 537.301 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 104.641 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 104.641 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 99.805 |
| 451.982 | 294.032 | 282.132 | -0.000 | 0.000 | -0.000 |
| 294.032 | 451.982 | 282.132 | 0.000 | 0.000 | -0.000 |
| 277.674 | 277.674 | 533.566 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 104.641 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 104.641 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 99.805 |
| 469.320 | 275.244 | 275.481 | -0.000 | -0.000 | 0.000 |
| 275.244 | 469.320 | 275.481 | -0.000 | -0.000 | 0.000 |
| 275.550 | 275.550 | 529.855 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 104.641 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 104.641 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 99.805 |
| 469.320 | 275.244 | 275.481 | 0.000 | 0.000 | -0.000 |
| 275.244 | 469.320 | 275.481 | 0.000 | -0.000 | -0.000 |
| 275.899 | 275.899 | 530.284 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 104.641 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 104.641 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 99.805 |
| 447.010 | 293.160 | 275.841 | -0.000 | -0.000 | 0.000 |
| 293.160 | 447.010 | 275.841 | -0.000 | -0.000 | 0.000 |
| 275.703 | 275.703 | 530.047 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 104.641 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 104.641 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 99.805 |
| 446.917 | 293.345 | 275.983 | 0.000 | 0.000 | -0.000 |
| 293.345 | 446.917 | 275.983 | 0.000 | 0.000 | -0.000 |
| 275.737 | 275.737 | 530.095 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 104.641 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 104.641 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 99.805 |
| 428.930 | 345.268 | 299.181 | -0.000 | -0.000 | -0.000 |
| 345.268 | 428.930 | 299.181 | -0.000 | 0.000 | 0.000 |
| 299.181 | 299.181 | 621.907 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 104.359 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 104.359 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 114.086 |
| 465.897 | 308.300 | 299.181 | 0.000 | 0.000 | -0.000 |
| 308.300 | 465.897 | 299.181 | 0.000 | 0.000 | 0.000 |
| 299.181 | 299.181 | 621.907 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 104.359 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 104.359 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 114.086 |
| 465.897 | 308.301 | 299.181 | -0.000 | -0.000 | -0.000 |
| 308.301 | 465.897 | 299.181 | -0.000 | -0.000 | -0.000 |
| 299.181 | 299.181 | 621.907 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 104.359 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 104.359 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 114.086 |
| 465.898 | 308.300 | 299.181 | 0.000 | -0.000 | -0.000 |
| 308.300 | 465.898 | 299.181 | -0.000 | 0.000 | 0.000 |
| 299.181 | 299.181 | 621.908 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 104.359 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 104.359 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 114.086 |
| 465.895 | 308.301 | 299.181 | -0.000 | -0.000 | 0.000 |
| 308.301 | 465.895 | 299.181 | -0.000 | -0.000 | -0.000 |
| 299.181 | 299.181 | 621.903 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 114.612 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 114.612 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 114.086 |
| 465.900 | 308.300 | 299.180 | 0.000 | 0.000 | -0.000 |
| 308.300 | 465.900 | 299.180 | 0.000 | 0.000 | 0.000 |
| 299.180 | 299.180 | 621.911 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 114.612 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 114.612 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 114.086 |
| 678.355 | 243.039 | 243.039 | -0.000 | -0.000 | -0.000 |
| 243.039 | 678.355 | 243.039 | 0.000 | -0.000 | -0.000 |
| 243.039 | 243.039 | 678.355 | 0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 94.120 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 94.120 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 94.120 |
| 678.355 | 243.039 | 243.039 | 0.000 | 0.000 | 0.000 |
| 243.039 | 678.355 | 243.039 | -0.000 | 0.000 | 0.000 |
| 243.039 | 243.039 | 678.355 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 94.120 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 94.120 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 94.120 |
| 678.354 | 243.039 | 243.039 | -0.000 | -0.000 | -0.000 |
| 243.039 | 678.354 | 243.039 | 0.000 | -0.000 | -0.000 |
| 243.039 | 243.039 | 678.354 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 94.120 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 94.120 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 94.120 |
| 678.355 | 243.039 | 243.039 | 0.000 | 0.000 | 0.000 |
| 243.039 | 678.355 | 243.039 | 0.000 | 0.000 | -0.000 |
| 243.039 | 243.039 | 678.355 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 94.120 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 94.120 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 94.120 |
| 678.351 | 243.039 | 243.039 | -0.000 | -0.000 | 0.000 |
| 243.039 | 678.351 | 243.039 | 0.000 | -0.000 | -0.000 |
| 243.039 | 243.039 | 678.351 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 94.120 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 94.120 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 94.120 |
| 678.359 | 243.039 | 243.039 | 0.000 | 0.000 | 0.000 |
| 243.039 | 678.359 | 243.039 | 0.000 | 0.000 | -0.000 |
| 243.039 | 243.039 | 678.359 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 94.120 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 94.120 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 94.120 |
| 137.504 | 112.709 | 112.709 | 0.000 | 0.000 | 0.000 |
| 112.709 | 137.504 | 112.709 | 0.000 | 0.000 | -0.000 |
| 112.709 | 112.709 | 137.504 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 112.574 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 112.574 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 112.574 |
| 137.504 | 112.709 | 112.709 | -0.000 | 0.000 | 0.000 |
| 112.709 | 137.504 | 112.709 | -0.000 | -0.000 | 0.000 |
| 112.709 | 112.709 | 137.504 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 112.574 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 112.574 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 112.574 |
| 137.504 | 112.709 | 112.709 | 0.000 | 0.000 | -0.000 |
| 112.709 | 137.504 | 112.709 | 0.000 | 0.000 | -0.000 |
| 112.709 | 112.709 | 137.504 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 112.574 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 112.574 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 112.574 |
| 137.504 | 112.709 | 112.709 | -0.000 | -0.000 | 0.000 |
| 112.709 | 137.504 | 112.709 | -0.000 | -0.000 | 0.000 |
| 112.709 | 112.709 | 137.504 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 112.574 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 112.574 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 112.574 |
| 137.503 | 112.709 | 112.709 | 0.000 | 0.000 | 0.000 |
| 112.709 | 137.503 | 112.709 | 0.000 | 0.000 | 0.000 |
| 112.709 | 112.709 | 137.503 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.001 | -0.000 | 112.574 | 0.000 | 0.000 |
| -0.001 | -0.000 | -0.000 | 0.000 | 112.574 | 0.000 |
| -0.001 | -0.000 | -0.000 | 0.000 | 0.000 | 112.574 |
| 137.505 | 112.710 | 112.710 | -0.000 | -0.000 | 0.000 |
| 112.710 | 137.505 | 112.710 | -0.000 | 0.000 | 0.000 |
| 112.710 | 112.710 | 137.505 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.001 | 0.000 | 112.574 | -0.000 | 0.000 |
| 0.001 | 0.000 | 0.000 | -0.000 | 112.574 | -0.000 |
| 0.001 | 0.000 | 0.000 | -0.000 | -0.000 | 112.574 |
| 115.931 | 30.620 | 30.620 | -0.000 | -0.000 | -0.000 |
| 30.620 | 115.931 | 30.620 | -0.000 | -0.000 | -0.000 |
| 30.620 | 30.620 | 115.931 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 30.550 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 30.550 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 30.550 |
| 115.931 | 30.620 | 30.620 | 0.000 | 0.000 | 0.000 |
| 30.620 | 115.931 | 30.620 | 0.000 | 0.000 | 0.000 |
| 30.620 | 30.620 | 115.931 | 0.000 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 30.550 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 30.550 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 30.550 |
| 115.931 | 30.620 | 30.620 | 0.000 | -0.000 | 0.000 |
| 30.620 | 115.931 | 30.620 | 0.000 | 0.000 | 0.000 |
| 30.620 | 30.620 | 115.931 | -0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 30.550 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 30.550 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 30.550 |
| 115.931 | 30.620 | 30.620 | -0.000 | -0.000 | -0.000 |
| 30.620 | 115.931 | 30.620 | 0.000 | 0.000 | -0.000 |
| 30.620 | 30.620 | 115.931 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 30.550 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 30.550 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 30.550 |
| 115.931 | 30.620 | 30.620 | -0.000 | -0.000 | 0.000 |
| 30.620 | 115.931 | 30.620 | 0.000 | -0.000 | 0.000 |
| 30.620 | 30.620 | 115.931 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 30.550 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 30.550 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 30.550 |
| 115.931 | 30.620 | 30.620 | -0.000 | 0.000 | 0.000 |
| 30.620 | 115.931 | 30.620 | 0.000 | 0.000 | 0.000 |
| 30.620 | 30.620 | 115.931 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 30.550 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 30.550 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 30.550 |
| 624.980 | 281.057 | 281.057 | -0.000 | 0.000 | 0.000 |
| 281.057 | 624.980 | 281.057 | -0.000 | 0.000 | -0.000 |
| 281.057 | 281.057 | 624.980 | 0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -108.286 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -108.286 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | -108.286 |
| 631.701 | 277.697 | 277.697 | -0.000 | -0.000 | -0.000 |
| 281.057 | 624.979 | 281.057 | 0.000 | -0.000 | -0.000 |
| 277.697 | 277.697 | 631.701 | 0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -108.286 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -108.286 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | -108.286 |
| 624.981 | 281.057 | 281.057 | -0.000 | -0.000 | 0.000 |
| 281.057 | 624.981 | 281.057 | -0.000 | -0.000 | 0.000 |
| 281.057 | 281.057 | 624.981 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 65.731 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 65.731 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 65.731 |
| 631.699 | 277.696 | 277.696 | -0.000 | 0.000 | -0.000 |
| 277.696 | 631.699 | 277.696 | -0.000 | 0.000 | -0.000 |
| 277.696 | 277.696 | 631.699 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 65.731 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 65.731 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 65.731 |
| 624.993 | 281.059 | 281.059 | -0.000 | -0.000 | 0.000 |
| 281.059 | 624.993 | 281.059 | -0.000 | 0.000 | 0.000 |
| 281.059 | 281.059 | 624.993 | -0.000 | -0.000 | 0.000 |
| 0.001 | 0.001 | 0.001 | 65.731 | 0.000 | -0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | 65.731 | -0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | -0.000 | 65.731 |
| 624.966 | 281.055 | 281.055 | -0.000 | 0.000 | -0.000 |
| 281.055 | 624.966 | 281.055 | 0.000 | -0.000 | -0.000 |
| 281.055 | 281.055 | 624.966 | 0.000 | 0.000 | -0.000 |
| -0.001 | -0.001 | -0.001 | 65.731 | -0.000 | -0.000 |
| -0.001 | -0.001 | -0.001 | 0.000 | 65.731 | 0.000 |
| -0.001 | -0.001 | -0.001 | -0.000 | -0.000 | 65.731 |
| -539848790.011 | -474351354.816 | -474099382.352 | 2324377.415 | 2846369.847 | -6652408.695 |
| -456888539.367 | -540717248.328 | -464356317.599 | 3032086.476 | 654424.293 | 2117257.682 |
| -461816837.195 | -467717103.976 | -528811577.143 | 2897057.391 | 324511.093 | 7050049.102 |
| 0.000 | 0.000 | 0.000 | 60.731 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 60.731 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 60.731 |
| 132.943 | 70.564 | 70.564 | -0.000 | -0.000 | 0.000 |
| 70.564 | 132.943 | 70.564 | -0.000 | -0.000 | 0.000 |
| 70.564 | 70.564 | 132.943 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 60.731 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 60.731 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 60.731 |
| -55464762.127 | -47006345.997 | -46584434.539 | -356095.554 | -62671.273 | -268330.785 |
| -45840513.978 | -54506634.492 | -47519024.828 | -675813.975 | -67974.235 | -169990.349 |
| -47154071.201 | -44269902.553 | -53641653.765 | -113660.691 | -417807.995 | 494505.901 |
| 0.000 | 0.000 | 0.000 | 60.731 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 60.731 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 60.731 |
| 45142801.890 | 50837023.157 | 49756514.625 | 388298.167 | 152269.326 | 161780.186 |
| 50003750.814 | 44942549.042 | 51606364.843 | -15043.837 | 937402.007 | 174324.032 |
| 51943605.926 | 50671218.134 | 45705946.447 | -285527.491 | 559523.181 | 291660.908 |
| -0.000 | -0.000 | -0.000 | 60.731 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 60.731 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 60.731 |
| -28287.253 | 5100893.401 | 5100893.435 | -0.000 | -0.000 | -0.000 |
| 5100893.435 | -28287.253 | 5100893.401 | -0.000 | 0.000 | -0.000 |
| 5100893.401 | 5100893.435 | -28287.253 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.001 | 0.001 | 66.343 | 0.000 | -0.000 |
| 0.001 | 0.000 | 0.001 | 0.000 | 66.343 | 0.000 |
| 0.001 | 0.001 | 0.000 | 0.000 | 0.000 | 66.343 |
| 4600865.583 | 5142797.059 | 5121056.730 | 33191.463 | -26683.562 | -21595.889 |
| 5069414.104 | 4572625.826 | 4997158.419 | -7392.023 | 46187.024 | -16083.781 |
| 5013606.829 | 4999203.475 | 4551857.023 | -25539.215 | 1937.945 | 52886.409 |
| -0.000 | -0.001 | -0.001 | 66.343 | -0.000 | 0.000 |
| -0.001 | -0.000 | -0.001 | -0.000 | 66.343 | 0.000 |
| -0.001 | -0.001 | -0.000 | 0.000 | 0.000 | 66.343 |
| 6.374 | 10.904 | 10.904 | 0.000 | 0.000 | -0.000 |
| 10.904 | 6.374 | 10.904 | 0.000 | 0.000 | -0.000 |
| 10.904 | 10.904 | 6.374 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.339 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.339 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 0.339 |
| 6.374 | 10.904 | 10.904 | -0.000 | -0.000 | -0.000 |
| 10.904 | 6.374 | 10.904 | -0.000 | -0.000 | 0.000 |
| 10.904 | 10.904 | 6.374 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.339 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.339 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 0.339 |
| 6.374 | 10.904 | 10.904 | 0.000 | 0.000 | 0.000 |
| 10.904 | 6.374 | 10.904 | -0.000 | 0.000 | -0.000 |
| 10.904 | 10.904 | 6.374 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.339 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.339 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 0.339 |
| 6515427.049 | 6131102.981 | 5945320.544 | -37456.546 | 13884.705 | -60568.386 |
| 6248425.524 | 6619482.041 | 5943407.394 | 56611.782 | 28017.938 | 1308.100 |
| 6111108.118 | 5943073.384 | 6648834.170 | 143097.304 | 6869.333 | -43460.976 |
| 0.000 | 0.000 | 0.000 | 0.339 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.339 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 0.339 |
| -466587.104 | -616512.379 | -629387.086 | -15.040 | -3464.906 | 6093.861 |
| -619218.886 | -493857.572 | -625168.004 | 1521.902 | -3148.558 | 5477.438 |
| -597872.889 | -616522.749 | -461880.441 | -11229.886 | -1123.123 | -3292.656 |
| -0.000 | -0.000 | -0.000 | 0.339 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.339 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.339 |
| 669058.869 | 600942.797 | 606217.904 | 621.840 | -3779.662 | 2328.606 |
| 602746.593 | 652439.657 | 583309.206 | 12123.279 | -4643.289 | -9186.611 |
| 611467.208 | 622991.243 | 657824.125 | -1633.066 | -4318.453 | 5565.598 |
| 0.000 | 0.000 | 0.000 | 0.339 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.339 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 0.339 |
| 562.354 | 626.683 | 626.683 | -0.000 | -0.000 | -0.000 |
| 626.683 | 562.354 | 626.683 | 0.000 | -0.000 | -0.000 |
| 626.683 | 626.683 | 562.354 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 338.744 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 338.744 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 338.744 |
| 562.353 | 626.683 | 626.683 | 0.000 | -0.000 | 0.000 |
| 626.683 | 562.353 | 626.683 | -0.000 | 0.000 | 0.000 |
| 626.683 | 626.683 | 562.353 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 338.744 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 338.744 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 338.744 |
| 562.355 | 626.683 | 626.683 | 0.000 | -0.000 | -0.000 |
| 626.683 | 562.355 | 626.683 | 0.000 | -0.000 | -0.000 |
| 626.683 | 626.683 | 562.355 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 338.744 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 338.744 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 338.744 |
| 562.352 | 626.682 | 626.682 | -0.000 | 0.000 | 0.000 |
| 626.682 | 562.352 | 626.682 | -0.000 | 0.000 | 0.000 |
| 626.682 | 626.682 | 562.352 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 338.744 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 338.744 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 338.744 |
| 562.367 | 626.688 | 626.688 | 0.000 | -0.000 | -0.000 |
| 626.688 | 562.367 | 626.688 | 0.000 | -0.000 | 0.000 |
| 626.688 | 626.688 | 562.367 | 0.000 | -0.000 | -0.000 |
| 0.001 | 0.003 | 0.003 | 338.744 | -0.000 | -0.000 |
| 0.003 | 0.001 | 0.003 | 0.000 | 338.744 | -0.000 |
| 0.003 | 0.003 | 0.001 | 0.000 | 0.000 | 338.744 |
| 562.340 | 626.678 | 626.678 | 0.000 | 0.000 | 0.000 |
| 626.678 | 562.340 | 626.678 | -0.000 | 0.000 | 0.000 |
| 626.678 | 626.678 | 562.340 | -0.000 | 0.000 | 0.000 |
| -0.001 | -0.003 | -0.003 | 338.744 | 0.000 | -0.000 |
| -0.003 | -0.001 | -0.003 | -0.000 | 338.744 | 0.000 |
| -0.003 | -0.003 | -0.001 | -0.000 | 0.000 | 338.744 |
| 184.820 | 64.126 | 64.126 | -0.000 | -0.000 | -0.000 |
| 64.126 | 184.820 | 64.126 | 0.000 | -0.000 | 0.000 |
| 64.126 | 64.126 | 184.820 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 64.126 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 64.126 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 64.126 |
| 184.820 | 64.126 | 64.126 | 0.000 | 0.000 | 0.000 |
| 64.126 | 184.820 | 64.126 | -0.000 | 0.000 | -0.000 |
| 64.126 | 64.126 | 184.820 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 64.126 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 64.126 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 64.126 |
| 184.820 | 64.126 | 64.126 | 0.000 | -0.000 | 0.000 |
| 64.126 | 184.820 | 64.126 | -0.000 | -0.000 | 0.000 |
| 64.126 | 64.126 | 184.820 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 64.126 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 64.126 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 64.126 |
| 184.820 | 64.126 | 64.126 | 0.000 | 0.000 | 0.000 |
| 64.126 | 184.820 | 64.126 | -0.000 | 0.000 | 0.000 |
| 64.126 | 64.126 | 184.820 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 64.126 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 64.126 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 64.126 |
| 184.819 | 64.126 | 64.126 | 0.000 | -0.000 | 0.000 |
| 64.126 | 184.819 | 64.126 | 0.000 | -0.000 | -0.000 |
| 64.126 | 64.126 | 184.819 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 64.126 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 64.126 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 64.126 |
| 184.820 | 64.126 | 64.126 | 0.000 | 0.000 | 0.000 |
| 64.126 | 184.820 | 64.126 | 0.000 | 0.000 | 0.000 |
| 64.126 | 64.126 | 184.820 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 64.126 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 64.126 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 64.126 |
| 622.652 | 316.365 | 387.159 | 0.000 | -0.000 | -0.000 |
| 316.365 | 622.652 | 387.159 | 0.000 | -0.000 | -0.000 |
| 387.159 | 387.159 | 535.765 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 206.073 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 206.073 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 153.143 |
| 622.838 | 316.179 | 387.159 | 0.000 | -0.000 | 0.000 |
| 316.179 | 622.838 | 387.159 | -0.000 | 0.000 | 0.000 |
| 387.159 | 387.159 | 535.765 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 206.073 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 206.073 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 153.143 |
| 622.652 | 316.365 | 387.159 | 0.000 | -0.000 | -0.000 |
| 316.179 | 622.838 | 387.159 | 0.000 | 0.000 | 0.000 |
| 387.159 | 387.159 | 535.767 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 206.073 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 206.073 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 153.330 |
| 622.839 | 316.179 | 387.159 | -0.000 | 0.000 | -0.000 |
| 316.365 | 622.652 | 387.159 | -0.000 | 0.000 | -0.000 |
| 387.158 | 387.158 | 535.763 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 206.073 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 206.073 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 153.330 |
| 622.650 | 316.365 | 387.159 | -0.000 | -0.000 | -0.000 |
| 316.179 | 622.835 | 387.159 | 0.000 | 0.000 | -0.000 |
| 387.160 | 387.160 | 535.784 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.001 | 206.073 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.001 | 0.000 | 206.073 | -0.000 |
| -0.001 | -0.000 | -0.000 | -0.000 | -0.000 | 153.330 |
| 622.841 | 316.180 | 387.158 | -0.000 | -0.000 | 0.000 |
| 316.179 | 622.841 | 387.158 | -0.000 | -0.000 | -0.000 |
| 387.157 | 387.157 | 535.746 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.001 | 206.073 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.001 | -0.000 | 206.073 | -0.000 |
| 0.001 | 0.000 | 0.000 | -0.000 | 0.000 | 153.330 |
| 160.709 | 142.417 | 44.884 | 0.000 | 0.000 | -0.000 |
| 142.417 | 160.709 | 44.884 | 0.000 | 0.000 | -0.000 |
| 44.884 | 44.884 | 148.161 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 44.565 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 44.565 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 9.146 |
| 227.269 | 75.858 | 44.884 | -0.000 | -0.000 | 0.000 |
| 142.417 | 160.709 | 44.884 | -0.000 | -0.000 | 0.000 |
| 44.884 | 44.884 | 148.161 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 44.565 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 44.565 | -0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 9.146 |
| 160.709 | 142.417 | 44.884 | 0.000 | 0.000 | -0.000 |
| 142.417 | 160.709 | 44.884 | 0.000 | 0.000 | -0.000 |
| 44.884 | 44.884 | 148.161 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 44.565 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 44.565 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 9.146 |
| 160.709 | 142.417 | 44.884 | -0.000 | -0.000 | 0.000 |
| 75.858 | 227.269 | 44.884 | -0.000 | -0.000 | 0.000 |
| 44.884 | 44.884 | 148.161 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | 44.565 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 44.565 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -0.000 | 9.146 |
| 160.706 | 142.418 | 44.884 | 0.000 | 0.000 | -0.000 |
| 142.417 | 160.707 | 44.884 | 0.000 | 0.000 | -0.000 |
| 44.884 | 44.884 | 148.162 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 44.565 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 44.565 | -0.000 |
| -0.000 | -0.001 | 0.000 | 0.000 | 0.000 | 9.146 |
| 160.712 | 142.417 | 44.884 | -0.000 | -0.000 | 0.000 |
| 142.418 | 160.711 | 44.884 | -0.000 | -0.000 | 0.000 |
| 44.884 | 44.884 | 148.160 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 44.565 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 44.565 | 0.000 |
| 0.000 | 0.001 | -0.000 | -0.000 | -0.000 | 9.146 |
| 102.982 | 35.498 | 38.690 | -0.000 | -0.000 | 0.000 |
| 34.655 | 103.825 | 38.690 | 0.000 | -0.000 | 0.000 |
| 38.690 | 38.690 | 99.790 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 38.620 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 38.620 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 33.742 |
| 103.825 | 34.655 | 38.690 | 0.000 | -0.000 | -0.000 |
| 35.498 | 102.982 | 38.690 | 0.000 | -0.000 | -0.000 |
| 38.690 | 38.690 | 99.790 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 38.620 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 38.620 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 33.742 |
| 102.982 | 35.498 | 38.690 | -0.000 | 0.000 | 0.000 |
| 34.655 | 103.825 | 38.690 | -0.000 | -0.000 | 0.000 |
| 38.690 | 38.690 | 99.790 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 38.620 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 38.620 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 33.742 |
| 102.982 | 35.498 | 38.690 | 0.000 | -0.000 | -0.000 |
| 34.655 | 103.825 | 38.690 | 0.000 | 0.000 | -0.000 |
| 38.690 | 38.690 | 99.790 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 38.620 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 38.620 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -0.000 | 33.742 |
| 102.982 | 35.498 | 38.690 | -0.000 | -0.000 | 0.000 |
| 35.498 | 102.982 | 38.690 | -0.000 | -0.000 | 0.000 |
| 38.690 | 38.690 | 99.790 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 38.620 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 38.620 | 0.000 |
| -0.000 | 0.000 | -0.000 | -0.000 | 0.000 | 33.742 |
| 103.825 | 34.655 | 38.690 | 0.000 | 0.000 | -0.000 |
| 34.655 | 103.825 | 38.690 | 0.000 | 0.000 | -0.000 |
| 38.690 | 38.690 | 99.790 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 38.620 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 38.620 | -0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | 0.000 | 33.742 |
| 709.351 | 357.910 | 502.338 | -0.000 | -0.000 | -0.000 |
| 354.502 | 712.759 | 502.338 | -0.000 | -0.000 | -0.000 |
| 502.338 | 502.338 | 1233.857 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 305.672 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 305.672 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 175.720 |
| 709.351 | 357.910 | 502.338 | 0.000 | 0.000 | 0.000 |
| 357.910 | 709.351 | 502.338 | 0.000 | 0.000 | -0.000 |
| 502.338 | 502.338 | 1233.857 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 305.672 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 305.672 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 175.720 |
| 709.351 | 357.910 | 502.338 | -0.000 | -0.000 | -0.000 |
| 357.910 | 709.351 | 502.338 | -0.000 | -0.000 | 0.000 |
| 502.338 | 502.338 | 1233.858 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 305.672 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | 305.672 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 179.129 |
| 712.760 | 354.502 | 502.338 | 0.000 | -0.000 | -0.000 |
| 357.910 | 709.351 | 502.338 | 0.000 | 0.000 | -0.000 |
| 502.338 | 502.338 | 1233.856 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 305.672 | 0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 305.672 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 179.129 |
| 709.348 | 357.910 | 502.338 | -0.000 | -0.000 | 0.000 |
| 357.910 | 709.348 | 502.338 | -0.000 | -0.000 | 0.000 |
| 502.339 | 502.339 | 1233.869 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.001 | 0.001 | 305.672 | -0.000 | -0.000 |
| -0.001 | -0.000 | 0.001 | -0.000 | 305.672 | 0.000 |
| -0.001 | -0.000 | -0.000 | -0.000 | -0.000 | 175.720 |
| 712.762 | 354.502 | 502.338 | 0.000 | 0.000 | 0.000 |
| 357.910 | 709.354 | 502.338 | 0.000 | 0.000 | -0.000 |
| 502.337 | 502.337 | 1233.846 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.001 | -0.001 | 305.672 | -0.000 | -0.000 |
| 0.001 | 0.000 | -0.001 | 0.000 | 305.672 | 0.000 |
| 0.001 | 0.000 | 0.000 | 0.000 | 0.000 | 175.720 |
| -363.501 | -5.084 | 345.062 | -0.000 | -0.000 | -0.000 |
| -5.084 | -363.501 | 345.062 | -0.000 | 0.000 | -0.000 |
| 345.061 | 345.061 | 415.066 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 221.390 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 221.390 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -179.209 |
| -363.501 | -5.084 | 345.062 | 0.000 | -0.000 | 0.000 |
| -48.711 | -319.874 | 345.062 | -0.000 | 0.000 | -0.000 |
| 345.061 | 345.061 | 415.066 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 221.390 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 221.390 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -179.209 |
| -319.874 | -48.711 | 345.062 | 0.000 | -0.000 | 0.000 |
| -5.084 | -363.501 | 345.062 | -0.000 | 0.000 | -0.000 |
| 345.062 | 345.062 | 415.067 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 221.390 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 221.390 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -179.209 |
| -319.875 | -48.711 | 345.062 | 0.000 | -0.000 | -0.000 |
| -48.711 | -319.875 | 345.062 | -0.000 | 0.000 | -0.000 |
| 345.061 | 345.061 | 415.065 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 221.390 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 221.390 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | -179.209 |
| -363.494 | -5.084 | 345.064 | -0.000 | 0.000 | 0.000 |
| -5.081 | -363.497 | 345.064 | -0.000 | -0.000 | -0.000 |
| 345.063 | 345.063 | 415.075 | 0.000 | 0.000 | -0.000 |
| 0.001 | 0.001 | 0.001 | 221.390 | 0.000 | -0.000 |
| 0.001 | 0.001 | 0.001 | -0.000 | 221.390 | -0.000 |
| 0.001 | 0.001 | 0.000 | -0.000 | 0.000 | -179.209 |
| -363.509 | -5.084 | 345.060 | 0.000 | -0.000 | 0.000 |
| -5.087 | -363.505 | 345.060 | 0.000 | -0.000 | 0.000 |
| 345.060 | 345.060 | 415.058 | 0.000 | -0.000 | 0.000 |
| -0.001 | -0.001 | -0.001 | 221.390 | 0.000 | 0.000 |
| -0.001 | -0.001 | -0.001 | 0.000 | 221.390 | 0.000 |
| -0.001 | -0.001 | -0.000 | 0.000 | 0.000 | -179.209 |
| 46.667 | 36.777 | -10.853 | -0.000 | -0.000 | 0.000 |
| 20.879 | 62.566 | -10.853 | -0.000 | -0.000 | -0.000 |
| -10.853 | -10.853 | -85.232 | 0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -10.837 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -10.837 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 18.031 |
| 61.951 | 21.493 | -10.853 | 0.000 | 0.000 | -0.000 |
| -32806452.416 | -32806407.990 | 0.526 | 0.007 | 0.000 | 0.000 |
| -10.853 | -10.853 | -85.232 | 0.000 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -10.837 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | -10.837 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 18.031 |
| 62.566 | 20.879 | -10.853 | -0.000 | -0.000 | -0.000 |
| 20.879 | 62.566 | -10.853 | -0.000 | -0.000 | -0.000 |
| -10.853 | -10.853 | -85.232 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -10.837 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -10.837 | 0.000 |
| -0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 20.844 |
| 61.951 | 21.493 | -10.853 | 0.000 | 0.000 | -0.000 |
| 20.879 | 62.566 | -10.853 | 0.000 | 0.000 | 0.000 |
| -10.853 | -10.853 | -85.232 | -0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | -10.837 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -10.837 | 0.000 |
| 0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 20.844 |
| 62.565 | 20.878 | -10.853 | -0.000 | -0.000 | -0.000 |
| 20.878 | 62.565 | -10.853 | -0.000 | -0.000 | -0.000 |
| -10.853 | -10.853 | -85.233 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -10.837 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | -10.837 | -0.000 |
| -869732.582 | -1022944.537 | -2236651.363 | -39304.959 | -24025.092 | -118131.281 |
| 62.566 | 20.879 | -10.853 | 0.000 | 0.000 | 0.000 |
| 21.493 | 61.952 | -10.853 | 0.000 | 0.000 | -0.000 |
| -10.853 | -10.853 | -85.232 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | -10.837 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | -10.837 | -0.000 |
| 849706.288 | 972260.124 | 2237506.851 | 31491.672 | -22785.226 | -126303.960 |
| 102.982 | 35.498 | 38.690 | 0.000 | 0.000 | -0.000 |
| 35.498 | 102.982 | 38.690 | 0.000 | 0.000 | -0.000 |
| 38.690 | 38.690 | 99.790 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 38.620 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 38.620 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 33.742 |
| 103.825 | 34.655 | 38.690 | 0.000 | 0.000 | 0.000 |
| 34.655 | 103.825 | 38.690 | -0.000 | -0.000 | -0.000 |
| 38.690 | 38.690 | 99.790 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 38.620 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 38.620 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 33.742 |
| 103.825 | 34.655 | 38.690 | -0.000 | 0.000 | 0.000 |
| 35.498 | 102.982 | 38.690 | 0.000 | 0.000 | -0.000 |
| 38.690 | 38.690 | 99.790 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 38.620 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 38.620 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 33.742 |
| 103.825 | 34.655 | 38.690 | 0.000 | -0.000 | 0.000 |
| 35.498 | 102.982 | 38.690 | -0.000 | -0.000 | 0.000 |
| 38.690 | 38.690 | 99.790 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 38.620 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 38.620 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 33.742 |
| 103.825 | 34.655 | 38.690 | -0.000 | 0.000 | 0.000 |
| 35.498 | 102.982 | 38.690 | 0.000 | -0.000 | -0.000 |
| 38.690 | 38.690 | 99.790 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 38.620 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 38.620 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 33.742 |
| 102.982 | 35.498 | 38.690 | -0.000 | 0.000 | 0.000 |
| 35.498 | 102.982 | 38.690 | -0.000 | -0.000 | 0.000 |
| 38.690 | 38.690 | 99.790 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 38.620 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 38.620 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 33.742 |
| 257.409 | 194.072 | 194.072 | -0.000 | -0.000 | -0.000 |
| 194.072 | 257.409 | 194.072 | -0.000 | -0.000 | -0.000 |
| 194.072 | 194.072 | 257.409 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 61.216 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 61.216 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 61.216 |
| 257.409 | 194.072 | 194.072 | 0.000 | 0.000 | 0.000 |
| 194.072 | 257.409 | 194.072 | 0.000 | 0.000 | 0.000 |
| 194.072 | 194.072 | 257.409 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 61.216 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 61.216 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 61.216 |
| 257.409 | 194.072 | 194.072 | -0.000 | -0.000 | -0.000 |
| 194.072 | 257.409 | 194.072 | -0.000 | -0.000 | -0.000 |
| 194.072 | 194.072 | 257.409 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 61.216 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 61.216 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 61.216 |
| 257.409 | 194.072 | 194.072 | -0.000 | 0.000 | 0.000 |
| 194.072 | 257.409 | 194.072 | 0.000 | -0.000 | 0.000 |
| 194.072 | 194.072 | 257.409 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 61.216 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 61.216 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 61.216 |
| 257.410 | 194.073 | 194.073 | -0.000 | 0.000 | 0.000 |
| 194.073 | 257.410 | 194.073 | 0.000 | -0.000 | 0.000 |
| 194.073 | 194.073 | 257.410 | 0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 61.216 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 61.216 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 61.216 |
| 257.409 | 194.071 | 194.071 | 0.000 | -0.000 | -0.000 |
| 194.071 | 257.409 | 194.071 | -0.000 | 0.000 | -0.000 |
| 194.071 | 194.071 | 257.409 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 61.216 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 61.216 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 61.216 |
| 77.839 | 40.537 | 40.537 | -0.000 | 0.000 | 0.000 |
| 40.537 | 77.839 | 40.537 | 0.000 | -0.000 | 0.000 |
| 40.537 | 40.537 | 77.839 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 40.535 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 40.535 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 40.535 |
| 77.839 | 40.537 | 40.537 | 0.000 | -0.000 | -0.000 |
| 40.537 | 77.839 | 40.537 | -0.000 | 0.000 | -0.000 |
| 40.537 | 40.537 | 77.839 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 32.439 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 32.439 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 32.439 |
| 77.839 | 40.537 | 40.537 | -0.000 | 0.000 | 0.000 |
| 40.537 | 77.839 | 40.537 | 0.000 | -0.000 | 0.000 |
| 40.537 | 40.537 | 77.839 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | 0.000 | 35.817 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 35.817 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | 0.000 | 35.817 |
| 77.839 | 40.537 | 40.537 | 0.000 | -0.000 | -0.000 |
| 40.537 | 77.839 | 40.537 | -0.000 | 0.000 | -0.000 |
| 40.537 | 40.537 | 77.839 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 40.535 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 40.535 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 40.535 |
| 77.839 | 40.537 | 40.537 | -0.000 | 0.000 | 0.000 |
| 40.537 | 77.839 | 40.537 | 0.000 | -0.000 | 0.000 |
| 40.537 | 40.537 | 77.839 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 30.021 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 30.021 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 30.021 |
| 77.839 | 40.537 | 40.537 | 0.000 | -0.000 | -0.000 |
| 40.537 | 77.839 | 40.537 | -0.000 | 0.000 | -0.000 |
| 40.537 | 40.537 | 77.839 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 30.021 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 30.021 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 30.021 |
| 8.871 | 8.871 | 8.871 | 0.000 | 0.000 | 0.000 |
| 8.871 | 8.871 | 8.871 | 0.000 | 0.000 | 0.000 |
| 8.871 | 8.871 | 8.871 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| 8.871 | 8.871 | 8.871 | -0.000 | -0.000 | -0.000 |
| 8.871 | 8.871 | 8.871 | -0.000 | -0.000 | -0.000 |
| 8.871 | 8.871 | 8.871 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 8.871 | 8.871 | 8.871 | 0.000 | 0.000 | 0.000 |
| 8.871 | 8.871 | 8.871 | 0.000 | 0.000 | 0.000 |
| 8.871 | 8.871 | 8.871 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| 8.871 | 8.871 | 8.871 | 0.000 | -0.000 | -0.000 |
| 8.871 | 8.871 | 8.871 | -0.000 | 0.000 | -0.000 |
| 8.871 | 8.871 | 8.871 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | -0.000 |
| 8.871 | 8.871 | 8.871 | 0.000 | 0.000 | 0.000 |
| 8.871 | 8.871 | 8.871 | 0.000 | 0.000 | 0.000 |
| 8.871 | 8.871 | 8.871 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 0.000 |
| 8.871 | 8.871 | 8.871 | -0.000 | -0.000 | -0.000 |
| 8.871 | 8.871 | 8.871 | -0.000 | -0.000 | -0.000 |
| 8.871 | 8.871 | 8.871 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 0.000 |
| 140.651 | 450.707 | 54.351 | 0.000 | 0.000 | 0.000 |
| 450.708 | 140.650 | 54.351 | 0.000 | 0.000 | 0.000 |
| 54.351 | 54.351 | 542.471 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | -64.093 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -64.093 | 0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 173.494 |
| 395.126 | 196.232 | 54.351 | -0.000 | 0.000 | -0.000 |
| 196.232 | 395.126 | 54.351 | -0.000 | 0.000 | -0.000 |
| 54.351 | 54.351 | 542.471 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -64.093 | 0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | -64.093 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 173.494 |
| 140.650 | 450.708 | 54.351 | -0.000 | -0.000 | 0.000 |
| 450.708 | 140.650 | 54.351 | 0.000 | -0.000 | 0.000 |
| 54.351 | 54.351 | 542.472 | 0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -64.093 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -64.093 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 0.000 | 173.494 |
| 140.651 | 450.707 | 54.351 | -0.000 | 0.000 | -0.000 |
| 450.707 | 140.651 | 54.351 | -0.000 | 0.000 | -0.000 |
| 54.351 | 54.351 | 542.471 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -64.093 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -64.093 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 173.494 |
| 140.645 | 450.709 | 54.352 | -0.000 | -0.000 | 0.000 |
| 450.709 | 140.645 | 54.352 | 0.000 | -0.000 | 0.000 |
| 54.351 | 54.351 | 542.476 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -64.093 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -64.093 | 0.000 |
| -0.001 | -0.000 | -0.000 | -0.000 | 0.000 | 173.494 |
| 140.656 | 450.706 | 54.351 | 0.000 | -0.000 | -0.000 |
| 450.706 | 140.656 | 54.351 | -0.000 | 0.000 | 0.000 |
| 54.351 | 54.351 | 542.467 | -0.000 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -64.093 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -64.093 | -0.000 |
| 0.001 | 0.000 | 0.000 | -0.000 | -0.000 | 173.494 |
| -64095099.714 | -228012382.652 | -266262450.359 | 3894334.055 | 5793452.783 | 5163073.610 |
| -245699228.650 | -94096011.674 | -280014607.427 | -13504831.280 | 1382518.050 | -1509952.879 |
| -73.323 | -73.323 | 0.893 | -0.000 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -73.327 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -73.327 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | -78.695 |
| 208185945.692 | 75019196.960 | 227622802.327 | 2091077.926 | -4136904.845 | -2616371.328 |
| 25636298.865 | 175800891.914 | 163707208.811 | -252903.726 | 3405864.912 | -137838.661 |
| -73.323 | -73.323 | 0.893 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -73.327 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -73.327 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | -78.695 |
| -6474829.036 | -17443337.268 | -16348899.649 | -169138.143 | 1070153.764 | -629331.282 |
| -15124265.343 | -1050271.310 | -13480958.828 | 34814.593 | 139609.673 | 5401.364 |
| -73.323 | -73.323 | 0.893 | -0.000 | 0.000 | 0.000 |
| -15888977.920 | -16562883.367 | -15393680.888 | 997465.057 | 693431.193 | 314575.316 |
| -23073458.800 | -23604315.689 | -18316606.905 | 177101.676 | 949744.225 | -118789.080 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -78.695 |
| 22085201.200 | 4322816.164 | 20448416.727 | -1961004.514 | 215094.312 | 200820.052 |
| 3653237.896 | 19360218.963 | 18689267.999 | 291351.656 | 118407.892 | -60547.613 |
| -73.323 | -73.323 | 0.893 | 0.000 | -0.000 | -0.000 |
| 18205985.319 | 15854694.249 | 15082713.194 | 461936.286 | -250031.944 | 360904.467 |
| 21600075.839 | 22195376.343 | 17392573.689 | -79958.550 | -127407.326 | -809855.169 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | -78.695 |
| -314956.517 | -2073627.089 | -2028477.131 | -65365.751 | -132128.651 | -64066.313 |
| -1993880.235 | -242554.706 | -2320728.018 | -70165.717 | 5642.259 | 10548.244 |
| -73.323 | -73.323 | 0.892 | -0.000 | 0.000 | 0.000 |
| -1849446.864 | -1639194.225 | -1433211.768 | 126291.071 | -38361.977 | 39722.517 |
| -2239164.420 | -2266441.974 | -1911250.297 | 36846.334 | -33203.518 | -54436.822 |
| -0.001 | -0.001 | -0.000 | -0.000 | 0.000 | -78.695 |
| 2089581.098 | 508259.301 | 1781490.833 | -119532.359 | 11738.603 | 5354.919 |
| 258812.475 | 2149952.737 | 2049027.911 | 6154.494 | 313925.400 | -2718.511 |
| -73.323 | -73.323 | 0.894 | 0.000 | -0.000 | -0.000 |
| 2134966.233 | 1836385.656 | 1557366.977 | 65153.666 | 10645.122 | 18239.075 |
| 2112829.269 | 2347272.694 | 1890563.850 | -12984.584 | 22596.621 | -21495.290 |
| 0.001 | 0.001 | 0.000 | 0.000 | -0.000 | -78.695 |
| 43.680 | 79.654 | 24.458 | 0.000 | -0.000 | 0.000 |
| 79.654 | 43.680 | 24.458 | -0.000 | -0.000 | 0.000 |
| 24.458 | 24.458 | 72.081 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -4.449 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -4.449 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 34.471 |
| 43.680 | 79.654 | 24.458 | -0.000 | 0.000 | -0.000 |
| 79.654 | 43.680 | 24.458 | 0.000 | -0.000 | -0.000 |
| 24.458 | 24.458 | 72.081 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -4.449 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -4.449 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 34.471 |
| 43.680 | 79.654 | 24.458 | 0.000 | 0.000 | 0.000 |
| 79.654 | 43.680 | 24.458 | 0.000 | -0.000 | 0.000 |
| 24.458 | 24.458 | 72.081 | 0.000 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -4.449 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -4.449 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -0.000 | 34.471 |
| 43.680 | 79.654 | 24.458 | -0.000 | 0.000 | -0.000 |
| 79.654 | 43.680 | 24.458 | -0.000 | 0.000 | -0.000 |
| 24.458 | 24.458 | 72.081 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -4.449 | -0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -4.449 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 34.471 |
| 43.680 | 79.653 | 24.458 | 0.000 | -0.000 | 0.000 |
| 79.653 | 43.680 | 24.458 | -0.000 | -0.000 | 0.000 |
| 24.458 | 24.458 | 72.081 | 0.000 | -0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | -4.449 | 0.000 | 0.000 |
| 0.000 | -0.000 | 0.000 | 0.000 | -4.449 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 34.471 |
| 43.679 | 79.654 | 24.458 | -0.000 | -0.000 | -0.000 |
| 79.654 | 43.679 | 24.458 | 0.000 | 0.000 | -0.000 |
| 24.458 | 24.458 | 72.081 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -4.449 | -0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -4.449 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 34.471 |
| 47.271 | 91.957 | 7.429 | 0.000 | 0.000 | 0.000 |
| 91.957 | 47.271 | 7.429 | 0.000 | 0.000 | 0.000 |
| 7.429 | 7.429 | 261.363 | 0.000 | 0.000 | -0.000 |
| -0.000 | 0.000 | -0.000 | -5.746 | 0.000 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -5.746 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 32.413 |
| 47.271 | 91.957 | 7.429 | -0.000 | -0.000 | -0.000 |
| 91.957 | 47.271 | 7.429 | -0.000 | -0.000 | -0.000 |
| 7.429 | 7.429 | 261.363 | -0.000 | -0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -5.746 | 0.000 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -5.746 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 32.413 |
| 33.111 | 106.116 | 7.429 | 0.000 | 0.000 | 0.000 |
| 106.116 | 33.111 | 7.429 | 0.000 | -0.000 | 0.000 |
| 7.429 | 7.429 | 261.363 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -5.746 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -5.746 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 32.413 |
| 47.271 | 91.957 | 7.429 | -0.000 | -0.000 | -0.000 |
| 91.957 | 47.271 | 7.429 | -0.000 | -0.000 | -0.000 |
| 7.429 | 7.429 | 261.363 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -5.746 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -5.746 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 32.413 |
| 33.110 | 106.117 | 7.428 | 0.000 | 0.000 | 0.000 |
| 106.117 | 33.110 | 7.428 | 0.000 | -0.000 | 0.000 |
| 7.429 | 7.429 | 261.365 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | -5.746 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | -5.746 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 32.413 |
| 47.272 | 91.957 | 7.429 | -0.000 | -0.000 | -0.000 |
| 91.957 | 47.272 | 7.429 | -0.000 | -0.000 | -0.000 |
| 7.429 | 7.429 | 261.362 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -5.746 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -5.746 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 32.413 |
| 317.783 | 108.343 | 73.460 | -20.967 | 0.000 | 0.000 |
| -4.699 | 194.823 | -200.006 | -0.684 | 0.000 | 0.000 |
| -208.163 | -208.163 | -556.279 | 0.000 | 0.000 | 0.000 |
| -20.967 | 20.967 | 0.000 | 76.597 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 76.597 | -20.967 |
| -0.000 | 0.000 | 0.000 | 0.000 | -20.967 | 104.720 |
| 317.783 | 108.343 | 73.460 | -20.967 | -0.000 | -0.000 |
| -5.455 | 194.061 | -201.765 | -0.697 | -0.000 | -0.000 |
| 80.525 | 80.525 | 112.752 | 0.000 | -0.000 | -0.000 |
| -11.342 | 11.030 | -0.362 | 33.889 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 76.597 | -20.967 |
| 0.000 | -0.000 | -0.000 | -0.000 | -20.967 | 104.720 |
| 317.783 | 108.343 | 73.460 | -20.967 | -0.000 | 0.000 |
| 108.343 | 317.783 | 73.460 | 20.967 | 0.000 | 0.000 |
| 80.525 | 80.525 | 112.752 | 0.000 | 0.000 | 0.000 |
| -20.967 | 20.967 | 0.000 | 76.597 | 0.000 | 0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 76.597 | -20.967 |
| -0.000 | -0.000 | 0.000 | 0.000 | -20.967 | 104.720 |
| 194.405 | -5.114 | -200.971 | 0.691 | 0.000 | -0.000 |
| -5.114 | 194.405 | -200.971 | -0.691 | -0.000 | -0.000 |
| -208.557 | -208.557 | -557.192 | -0.000 | -0.000 | -0.000 |
| -11.202 | 11.171 | -0.036 | 33.889 | 0.000 | -0.000 |
| 0.000 | -0.000 | -0.000 | -0.000 | 76.597 | -20.967 |
| 0.000 | 0.000 | -0.000 | -0.000 | -20.967 | 104.720 |
| 190.809 | -14.148 | -215.606 | -11.185 | 0.000 | 0.000 |
| -14.148 | 190.809 | -215.606 | 11.185 | 0.000 | 0.000 |
| -208.581 | -208.581 | -557.260 | 0.000 | 0.000 | 0.000 |
| -20.967 | 20.967 | -0.000 | 76.597 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 76.597 | -20.967 |
| 0.001 | 0.001 | 0.004 | 0.000 | -11.186 | 102.480 |
| 194.443 | -5.079 | -200.889 | 0.691 | 0.000 | -0.000 |
| 108.344 | 317.786 | 73.460 | 20.967 | 0.000 | -0.000 |
| -208.472 | -208.472 | -556.984 | -0.000 | -0.000 | -0.000 |
| -20.967 | 20.967 | 0.000 | 76.597 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 76.597 | -20.967 |
| -0.001 | -0.001 | -0.004 | -0.000 | -11.186 | 102.480 |
| 288.784 | 81.259 | 81.259 | -0.000 | -0.000 | 0.000 |
| 81.259 | 288.784 | 81.259 | -0.000 | -0.000 | -0.000 |
| 81.259 | 81.259 | 288.784 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 42.399 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 42.399 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 42.399 |
| 288.784 | 81.259 | 81.259 | 0.000 | 0.000 | 0.000 |
| 81.259 | 288.784 | 81.259 | 0.000 | 0.000 | 0.000 |
| 81.259 | 81.259 | 288.784 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 42.399 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 42.399 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 42.399 |
| 288.785 | 81.259 | 81.259 | -0.000 | -0.000 | 0.000 |
| 81.259 | 288.785 | 81.259 | -0.000 | -0.000 | 0.000 |
| 81.259 | 81.259 | 288.785 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 42.399 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 42.399 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 42.399 |
| 288.783 | 81.259 | 81.259 | 0.000 | 0.000 | 0.000 |
| 81.259 | 288.783 | 81.259 | 0.000 | 0.000 | 0.000 |
| 81.259 | 81.259 | 288.783 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 42.399 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 42.399 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 42.399 |
| 288.791 | 81.261 | 81.261 | -0.000 | -0.000 | 0.000 |
| 81.261 | 288.791 | 81.261 | -0.000 | -0.000 | 0.000 |
| 81.261 | 81.261 | 288.791 | -0.000 | -0.000 | 0.000 |
| 0.001 | 0.002 | 0.002 | 42.399 | -0.000 | -0.000 |
| 0.002 | 0.001 | 0.002 | -0.000 | 42.399 | 0.000 |
| 0.002 | 0.002 | 0.001 | -0.000 | -0.000 | 42.399 |
| 288.777 | 81.257 | 81.257 | 0.000 | 0.000 | -0.000 |
| 81.257 | 288.777 | 81.257 | 0.000 | 0.000 | -0.000 |
| 81.257 | 81.257 | 288.777 | 0.000 | -0.000 | -0.000 |
| -0.001 | -0.002 | -0.002 | 42.399 | 0.000 | -0.000 |
| -0.002 | -0.001 | -0.002 | 0.000 | 42.399 | -0.000 |
| -0.002 | -0.002 | -0.001 | 0.000 | 0.000 | 42.399 |
| 266.820 | 47.869 | 47.869 | 0.000 | 0.000 | -0.000 |
| 47.869 | 266.820 | 47.869 | 0.000 | 0.000 | 0.000 |
| 47.869 | 47.869 | 266.820 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 25.225 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 25.225 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 25.225 |
| 266.820 | 47.869 | 47.869 | -0.000 | -0.000 | 0.000 |
| 47.869 | 266.820 | 47.869 | -0.000 | -0.000 | 0.000 |
| 47.869 | 47.869 | 266.820 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 25.225 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 25.225 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | -0.000 | 25.225 |
| 266.820 | 47.869 | 47.869 | 0.000 | 0.000 | 0.000 |
| 47.869 | 266.820 | 47.869 | 0.000 | 0.000 | 0.000 |
| 47.869 | 47.869 | 266.820 | 0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 25.225 | 0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 25.225 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 25.225 |
| 266.821 | 47.869 | 47.869 | -0.000 | -0.000 | -0.000 |
| 47.869 | 266.821 | 47.869 | -0.000 | -0.000 | -0.000 |
| 47.869 | 47.869 | 266.821 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 25.225 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 25.225 | -0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | -0.000 | 25.225 |
| 266.815 | 47.868 | 47.868 | 0.000 | 0.000 | -0.000 |
| 47.868 | 266.815 | 47.868 | 0.000 | 0.000 | 0.000 |
| 47.868 | 47.868 | 266.815 | 0.000 | 0.000 | 0.000 |
| -0.001 | -0.002 | -0.002 | 25.225 | 0.000 | 0.000 |
| -0.002 | -0.001 | -0.002 | 0.000 | 25.225 | 0.000 |
| -0.002 | -0.002 | -0.001 | 0.000 | 0.000 | 25.225 |
| 266.826 | 47.871 | 47.871 | -0.000 | -0.000 | -0.000 |
| 47.871 | 266.826 | 47.871 | -0.000 | -0.000 | -0.000 |
| 47.871 | 47.871 | 266.826 | 0.000 | -0.000 | -0.000 |
| 0.001 | 0.002 | 0.002 | 25.225 | -0.000 | 0.000 |
| 0.002 | 0.001 | 0.002 | -0.000 | 25.225 | 0.000 |
| 0.002 | 0.002 | 0.001 | -0.000 | -0.000 | 25.225 |
| 98.381 | 25.091 | 25.091 | -0.000 | -0.000 | -0.000 |
| 25.091 | 98.381 | 25.091 | -0.000 | -0.000 | -0.000 |
| 25.091 | 25.091 | 98.381 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 25.068 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 25.068 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 25.068 |
| 98.381 | 25.091 | 25.091 | 0.000 | 0.000 | 0.000 |
| 25.091 | 98.381 | 25.091 | 0.000 | 0.000 | 0.000 |
| 25.091 | 25.091 | 98.381 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 25.068 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 25.068 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 25.068 |
| 98.381 | 25.091 | 25.091 | -0.000 | -0.000 | -0.000 |
| 25.091 | 98.381 | 25.091 | -0.000 | -0.000 | -0.000 |
| 25.091 | 25.091 | 98.381 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 25.068 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 25.068 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 25.068 |
| 98.382 | 25.091 | 25.091 | 0.000 | 0.000 | 0.000 |
| 25.091 | 98.382 | 25.091 | 0.000 | 0.000 | 0.000 |
| 25.091 | 25.091 | 98.382 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 25.068 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 25.068 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 25.068 |
| 98.381 | 25.091 | 25.091 | -0.000 | -0.000 | -0.000 |
| 25.091 | 98.381 | 25.091 | -0.000 | -0.000 | -0.000 |
| 25.091 | 25.091 | 98.381 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 25.068 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 25.068 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 25.068 |
| 98.382 | 25.091 | 25.091 | 0.000 | 0.000 | 0.000 |
| 25.091 | 98.382 | 25.091 | 0.000 | 0.000 | 0.000 |
| 25.091 | 25.091 | 98.382 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 25.068 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 25.068 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 25.068 |
| 457.961 | 273.252 | 211.406 | 0.000 | 0.000 | 0.000 |
| 282.003 | 732.390 | 281.988 | -0.000 | 0.000 | 0.000 |
| 211.402 | 273.233 | 457.761 | -0.000 | 0.000 | 0.000 |
| 0.082 | 0.013 | 0.082 | -7.481 | 0.000 | 0.000 |
| 0.045 | 0.007 | 0.045 | 0.000 | 127.070 | 0.000 |
| 0.082 | 0.013 | 0.082 | 0.000 | 0.000 | 1.314 |
| 375.539 | 260.398 | 128.981 | 0.000 | -0.000 | -0.000 |
| 269.128 | 730.383 | 269.112 | -0.000 | -0.000 | -0.000 |
| 128.977 | 260.378 | 375.332 | 0.000 | -0.000 | -0.000 |
| -0.082 | -0.013 | -0.082 | -7.481 | -0.000 | -0.000 |
| -0.044 | -0.007 | -0.045 | -0.000 | 127.070 | -0.000 |
| -0.082 | -0.013 | -0.082 | 0.000 | -0.000 | 1.314 |
| 457.961 | 273.252 | 211.406 | 0.000 | 0.000 | 0.000 |
| 282.003 | 732.390 | 281.988 | -0.000 | 0.000 | 0.000 |
| 129.003 | 260.382 | 375.357 | 0.000 | -0.000 | 0.000 |
| 0.008 | 0.001 | 0.008 | -7.482 | 0.000 | -0.000 |
| 0.004 | 0.001 | 0.004 | 0.000 | 127.070 | 0.000 |
| 0.008 | 0.001 | 0.008 | -0.000 | 0.000 | 1.312 |
| 375.560 | 260.401 | 129.002 | 0.000 | -0.000 | -0.000 |
| 269.150 | 730.386 | 269.134 | 0.000 | -0.000 | -0.000 |
| 128.998 | 260.381 | 375.354 | 0.000 | -0.000 | -0.000 |
| -0.008 | -0.001 | -0.008 | -7.482 | -0.000 | -0.000 |
| -0.004 | -0.001 | -0.004 | -0.000 | 127.070 | -0.000 |
| -0.008 | -0.001 | -0.008 | 0.000 | -0.000 | 1.312 |
| 375.558 | 260.401 | 129.006 | -0.000 | 0.000 | 0.000 |
| 269.153 | 730.383 | 269.137 | -0.000 | 0.000 | 0.000 |
| 129.002 | 260.381 | 375.352 | -0.000 | 0.000 | 0.000 |
| -0.001 | -0.002 | -0.002 | -7.482 | 0.000 | -0.000 |
| -0.001 | -0.001 | -0.001 | -0.000 | 127.070 | -0.000 |
| -0.001 | -0.001 | -0.001 | -0.000 | 0.000 | 188.362 |
| 457.967 | 273.253 | 211.406 | -0.000 | 0.000 | -0.000 |
| 269.152 | 730.389 | 269.136 | 0.000 | -0.000 | -0.000 |
| 128.998 | 260.382 | 375.359 | -0.000 | -0.000 | -0.000 |
| 0.001 | 0.002 | 0.002 | -7.482 | -0.000 | -0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | 127.070 | -0.000 |
| 0.001 | 0.001 | 0.001 | 0.000 | -0.000 | 188.362 |
| 15214.035 | 15176.117 | 15176.117 | -0.000 | -0.000 | -0.000 |
| 15176.117 | 15214.035 | 15176.117 | -0.000 | -0.000 | -0.000 |
| 15176.117 | 15176.117 | 15214.035 | -0.000 | -0.000 | -0.000 |
| 3081.487 | 3081.487 | 3081.487 | 24.029 | 0.000 | 0.000 |
| 3081.487 | 3081.487 | 3081.487 | 0.000 | 24.029 | -0.000 |
| 3081.487 | 3081.487 | 3081.487 | -0.000 | -0.000 | 24.029 |
| -2718.431 | -2769.244 | -2769.244 | -0.000 | -0.000 | -0.000 |
| -2769.244 | -2718.431 | -2769.244 | -0.000 | -0.000 | -0.000 |
| -2769.244 | -2769.244 | -2718.431 | -0.000 | -0.000 | -0.000 |
| -3081.487 | -3081.487 | -3081.487 | 24.029 | 0.000 | 0.000 |
| -3081.487 | -3081.487 | -3081.487 | 0.000 | 24.029 | -0.000 |
| -3081.487 | -3081.487 | -3081.487 | -0.000 | 0.000 | 24.029 |
| 494.095 | 278.126 | 278.126 | -0.000 | 0.000 | 0.000 |
| 278.126 | 494.095 | 278.126 | -0.000 | -0.000 | 0.000 |
| 278.126 | 278.126 | 494.095 | 0.000 | 0.000 | -0.000 |
| 1068.875 | 1068.875 | 1068.875 | 85.436 | -0.000 | -0.000 |
| 1068.875 | 1068.875 | 1068.875 | -0.000 | 85.436 | 0.000 |
| 1068.875 | 1068.875 | 1068.875 | -0.000 | 0.000 | 85.436 |
| -998.510 | -1041.767 | -1041.767 | 0.000 | 0.000 | 0.000 |
| -1041.767 | -998.510 | -1041.767 | 0.000 | -0.000 | 0.000 |
| -1041.767 | -1041.767 | -998.510 | 0.000 | 0.000 | 0.000 |
| -1068.875 | -1068.875 | -1068.875 | 85.436 | -0.000 | 0.000 |
| -1068.875 | -1068.875 | -1068.875 | -0.000 | 85.436 | 0.000 |
| -1068.875 | -1068.875 | -1068.875 | -0.000 | -0.000 | 85.436 |
| 451.711 | 386.741 | 386.741 | -0.000 | -0.000 | -0.000 |
| 386.741 | 451.711 | 386.741 | -0.000 | -0.000 | -0.000 |
| 386.741 | 386.741 | 451.711 | -0.000 | -0.000 | -0.000 |
| 132.159 | 132.159 | 132.159 | 24.955 | -0.000 | -0.000 |
| 132.159 | 132.159 | 132.159 | -0.000 | 24.955 | -0.000 |
| 132.159 | 132.159 | 132.159 | -0.000 | -0.000 | 24.955 |
| 494.092 | 278.125 | 278.125 | -0.000 | -0.000 | -0.000 |
| 278.125 | 494.092 | 278.125 | -0.000 | -0.000 | 0.000 |
| 278.125 | 278.125 | 494.092 | -0.000 | -0.000 | -0.000 |
| -132.159 | -132.159 | -132.159 | 24.955 | 0.000 | 0.000 |
| -132.159 | -132.159 | -132.159 | 0.000 | 24.955 | 0.000 |
| -132.159 | -132.159 | -132.159 | -0.000 | 0.000 | 24.955 |
| 348.943 | 266.807 | 266.807 | 0.000 | -0.000 | -0.000 |
| 266.807 | 348.943 | 266.807 | -0.000 | 0.000 | -0.000 |
| 266.807 | 266.807 | 348.943 | 0.000 | 0.000 | -0.000 |
| -13.126 | -13.126 | -13.126 | 66.341 | 0.000 | 0.000 |
| -13.126 | -13.126 | -13.126 | 0.000 | 66.341 | 0.000 |
| -13.126 | -13.126 | -13.126 | 0.000 | 0.000 | 66.341 |
| 487.400 | 274.540 | 274.540 | -0.000 | -0.000 | 0.000 |
| 274.540 | 487.400 | 274.540 | 0.000 | -0.000 | 0.000 |
| 274.540 | 274.540 | 487.400 | -0.000 | -0.000 | -0.000 |
| 13.126 | 13.126 | 13.126 | 66.341 | -0.000 | -0.000 |
| 13.126 | 13.126 | 13.126 | -0.000 | 66.341 | -0.000 |
| 13.126 | 13.126 | 13.126 | 0.000 | -0.000 | 66.341 |
| 406.833 | 295.088 | 295.088 | 0.000 | 0.000 | 0.000 |
| 295.088 | 406.833 | 295.088 | 0.000 | 0.000 | 0.000 |
| 295.088 | 295.088 | 406.833 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 107.489 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 107.489 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 107.489 |
| 487.399 | 274.540 | 274.540 | 0.000 | -0.000 | 0.000 |
| 274.540 | 487.399 | 274.540 | 0.000 | 0.000 | -0.000 |
| 274.540 | 274.540 | 487.399 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 107.489 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 107.489 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 107.489 |
| 405.144 | 297.497 | 297.497 | 0.000 | 0.000 | 0.000 |
| 297.497 | 405.144 | 297.497 | 0.000 | 0.000 | 0.000 |
| 297.497 | 297.497 | 405.144 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 107.489 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 107.489 | 0.000 |
| 0.000 | 0.000 | 0.000 | -0.000 | 0.000 | 107.489 |
| 363.284 | 293.898 | 293.898 | -0.000 | -0.000 | -0.000 |
| 293.898 | 363.284 | 293.898 | -0.000 | -0.000 | -0.000 |
| 293.898 | 293.898 | 363.284 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 107.489 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | 107.489 | 0.000 |
| -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | 107.489 |
| 258.729 | 217.255 | 217.255 | -0.000 | -0.000 | 0.000 |
| 217.255 | 258.729 | 217.255 | 0.000 | -0.000 | 0.000 |
| 217.255 | 217.255 | 258.729 | -0.000 | -0.000 | 0.000 |
| -0.444 | -0.444 | -0.444 | 59.691 | -0.000 | 0.000 |
| -0.444 | -0.444 | -0.444 | -0.000 | 59.691 | 0.000 |
| -0.444 | -0.444 | -0.444 | -0.000 | -0.000 | 59.691 |
| 444.448 | 329.092 | 329.092 | 0.000 | -0.000 | 0.000 |
| 329.092 | 444.448 | 329.092 | 0.000 | -0.000 | 0.000 |
| 329.092 | 329.092 | 444.448 | 0.000 | -0.000 | 0.000 |
| 0.444 | 0.444 | 0.444 | 59.691 | 0.000 | -0.000 |
| 0.444 | 0.444 | 0.444 | 0.000 | 59.691 | -0.000 |
| 0.444 | 0.444 | 0.444 | 0.000 | 0.000 | 59.691 |
| 444.449 | 329.092 | 329.092 | -0.000 | 0.000 | -0.000 |
| 329.092 | 444.449 | 329.092 | -0.000 | 0.000 | -0.000 |
| 329.092 | 329.092 | 444.449 | -0.000 | 0.000 | -0.000 |
| -0.017 | -0.017 | -0.017 | 71.122 | 0.000 | -0.000 |
| -0.017 | -0.017 | -0.017 | -0.000 | 71.122 | 0.000 |
| -0.017 | -0.017 | -0.017 | -0.000 | 0.000 | 71.122 |
| 249.177 | 217.909 | 217.909 | 0.000 | 0.000 | -0.000 |
| 217.909 | 249.177 | 217.909 | -0.000 | 0.000 | -0.000 |
| 217.909 | 217.909 | 249.177 | 0.000 | 0.000 | -0.000 |
| 0.017 | 0.017 | 0.017 | 71.122 | -0.000 | -0.000 |
| 0.017 | 0.017 | 0.017 | -0.000 | 71.122 | -0.000 |
| 0.017 | 0.017 | 0.017 | 0.000 | -0.000 | 71.122 |
| 444.446 | 329.087 | 329.087 | -0.000 | 0.000 | -0.000 |
| 329.087 | 444.446 | 329.087 | -0.000 | 0.000 | -0.000 |
| 329.087 | 329.087 | 444.446 | 0.000 | 0.000 | -0.000 |
| -0.009 | -0.009 | -0.009 | 56.303 | 0.000 | 0.000 |
| -0.009 | -0.009 | -0.009 | -0.000 | 56.303 | 0.000 |
| -0.009 | -0.009 | -0.009 | -0.000 | 0.000 | 56.303 |
| 368.706 | 296.344 | 296.344 | 0.000 | -0.000 | -0.000 |
| 296.344 | 368.706 | 296.344 | -0.000 | -0.000 | -0.000 |
| 296.344 | 296.344 | 368.706 | 0.000 | -0.000 | -0.000 |
| 0.009 | 0.009 | 0.009 | 56.303 | -0.000 | -0.000 |
| 0.009 | 0.009 | 0.009 | 0.000 | 56.303 | -0.000 |
| 0.009 | 0.009 | 0.009 | 0.000 | -0.000 | 56.303 |
| 316.294 | 255.594 | 255.594 | -0.000 | -0.000 | 0.000 |
| 255.594 | 316.294 | 255.594 | 0.000 | 0.000 | 0.000 |
| 255.594 | 255.594 | 316.294 | 0.000 | -0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 112.355 | 0.000 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 112.355 | 0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 112.355 |
| 316.294 | 255.594 | 255.594 | -0.000 | -0.000 | -0.000 |
| 255.594 | 316.294 | 255.594 | -0.000 | -0.000 | -0.000 |
| 255.594 | 255.594 | 316.294 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 112.355 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 112.355 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 0.000 | 112.355 |
| -1169.161 | -1214.440 | -1214.440 | -0.000 | 0.000 | -0.000 |
| -1214.440 | -1169.161 | -1214.440 | -0.000 | 0.000 | -0.000 |
| -1214.440 | -1214.440 | -1169.161 | -0.000 | 0.000 | -0.000 |
| -1346.009 | -1346.009 | -1346.009 | 50.788 | 0.000 | -0.000 |
| -1346.010 | -1346.010 | -1346.010 | -0.000 | 50.788 | -0.000 |
| -1346.010 | -1346.010 | -1346.010 | -0.000 | 0.000 | 50.788 |
| 316.294 | 255.594 | 255.594 | -0.000 | -0.000 | -0.000 |
| 255.594 | 316.294 | 255.594 | -0.000 | 0.000 | -0.000 |
| 255.594 | 255.594 | 316.294 | -0.000 | 0.000 | -0.000 |
| 1346.010 | 1346.010 | 1346.010 | 50.788 | -0.000 | 0.000 |
| 1346.009 | 1346.009 | 1346.009 | 0.000 | 50.788 | 0.000 |
| 1346.009 | 1346.009 | 1346.009 | 0.000 | -0.000 | 50.788 |
| 233.298 | 220.413 | 220.413 | 0.000 | -0.000 | 0.000 |
| 220.413 | 233.298 | 220.413 | 0.000 | -0.000 | 0.000 |
| 220.413 | 220.413 | 233.298 | 0.000 | -0.000 | 0.000 |
| 8.694 | 8.694 | 8.694 | 89.501 | -0.000 | 0.000 |
| 8.694 | 8.694 | 8.694 | 0.000 | 89.501 | 0.000 |
| 8.694 | 8.694 | 8.694 | 0.000 | -0.000 | 89.501 |
| 318.298 | 276.882 | 276.882 | 0.000 | -0.000 | 0.000 |
| 276.882 | 318.298 | 276.882 | 0.000 | -0.000 | 0.000 |
| 276.882 | 276.882 | 318.298 | 0.000 | -0.000 | 0.000 |
| -8.694 | -8.694 | -8.694 | 89.501 | 0.000 | -0.000 |
| -8.694 | -8.694 | -8.694 | -0.000 | 89.501 | -0.000 |
| -8.694 | -8.694 | -8.694 | -0.000 | 0.000 | 89.501 |
| 950.623 | 287.135 | 295.875 | -0.000 | -0.000 | 0.000 |
| 248.601 | 459.557 | 125.142 | 0.000 | 0.000 | 0.000 |
| 223.967 | 116.796 | 28.352 | -0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 49.613 | 0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 0.000 | 158.359 | -0.000 |
| 9.114 | 7.033 | 10.866 | 0.000 | 0.000 | -584.783 |
| 950.623 | 287.135 | 295.875 | -0.000 | -0.000 | -0.000 |
| 294.455 | 494.939 | 179.809 | -0.000 | -0.000 | -0.000 |
| 294.969 | 171.584 | 113.002 | 0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | 49.613 | -0.000 | 0.000 |
| -0.000 | -0.000 | -0.000 | 0.000 | 158.359 | -0.000 |
| -9.114 | -7.033 | -10.866 | -0.000 | -0.000 | -584.783 |
| 890.856 | 241.017 | 224.619 | 0.000 | -0.000 | 0.000 |
| 294.455 | 494.939 | 179.809 | 0.000 | 0.000 | -0.000 |
| 223.709 | 116.597 | 28.045 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.000 | -0.000 | 49.613 | 0.000 | -0.000 |
| 0.029 | 0.022 | 0.034 | 0.000 | 158.359 | -0.000 |
| -27.196 | -20.986 | -32.423 | -0.000 | -0.000 | -869.586 |
| 950.624 | 287.135 | 295.874 | 0.000 | -0.000 | 0.000 |
| 294.455 | 494.939 | 179.809 | -0.000 | -0.000 | -0.000 |
| 294.969 | 171.584 | 113.002 | 0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | 0.000 | 49.613 | 0.000 | 0.000 |
| -0.029 | -0.022 | -0.034 | -0.000 | 158.359 | 0.000 |
| 27.196 | 20.986 | 32.423 | 0.000 | 0.000 | -869.586 |
| 890.826 | 240.997 | 224.589 | -0.000 | 0.000 | -0.000 |
| 294.456 | 494.941 | 179.809 | -0.000 | 0.000 | -0.000 |
| 223.685 | 116.578 | 28.017 | -0.000 | 0.000 | -0.000 |
| -0.000 | -0.001 | -0.000 | 49.613 | -0.000 | -0.000 |
| 0.001 | 0.000 | 0.002 | -0.000 | 158.359 | 0.000 |
| -2.849 | -2.209 | -3.390 | -0.000 | -0.000 | -869.586 |
| 890.828 | 240.993 | 224.581 | -0.000 | -0.000 | 0.000 |
| 294.454 | 494.937 | 179.809 | -0.000 | -0.000 | 0.000 |
| 223.676 | 116.574 | 28.005 | -0.000 | -0.000 | 0.000 |
| 0.000 | 0.001 | 0.000 | 49.613 | -0.000 | 0.000 |
| -0.001 | -0.000 | -0.002 | 0.000 | 158.359 | 0.000 |
| 2.849 | 2.209 | 3.390 | 0.000 | -0.000 | -869.586 |
| 684.843 | 349.629 | 490.526 | 0.000 | -0.000 | 0.000 |
| 347.911 | 701.365 | 493.027 | 0.000 | 0.000 | 0.000 |
| 494.447 | 495.621 | 1146.780 | -0.000 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 297.337 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | -0.000 | 295.949 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 166.860 |
| 684.843 | 349.629 | 490.526 | -0.000 | 0.000 | 0.000 |
| 350.644 | 698.808 | 493.319 | -0.000 | 0.000 | 0.000 |
| 494.135 | 495.912 | 1146.747 | -0.000 | -0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 297.337 | 0.000 | 0.000 |
| 0.000 | -0.000 | -0.000 | 0.000 | 295.949 | -0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | -0.000 | 166.860 |
| 687.765 | 346.896 | 490.838 | -0.000 | -0.000 | -0.000 |
| 350.644 | 698.808 | 493.319 | 0.000 | -0.000 | -0.000 |
| 494.447 | 495.621 | 1146.782 | 0.000 | 0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 297.337 | -0.000 | 0.000 |
| -0.000 | -0.000 | 0.000 | 0.000 | 295.949 | 0.000 |
| -0.000 | 0.000 | -0.000 | 0.000 | -0.000 | 166.860 |
| 684.843 | 349.629 | 490.526 | -0.000 | -0.000 | 0.000 |
| 350.644 | 698.808 | 493.319 | -0.000 | 0.000 | 0.000 |
| 494.135 | 495.912 | 1146.746 | -0.000 | -0.000 | -0.000 |
| 0.000 | 0.000 | -0.000 | 297.337 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 295.949 | 0.000 |
| 0.000 | -0.000 | 0.000 | -0.000 | 0.000 | 166.860 |
| 684.842 | 349.629 | 490.527 | 0.000 | -0.000 | -0.000 |
| 350.645 | 698.806 | 493.320 | 0.000 | -0.000 | 0.000 |
| 494.449 | 495.622 | 1146.795 | 0.000 | -0.000 | -0.000 |
| -0.000 | -0.000 | 0.001 | 297.343 | -0.000 | 0.000 |
| -0.001 | -0.000 | 0.001 | 0.000 | 295.949 | 0.000 |
| -0.001 | -0.000 | -0.000 | 0.000 | -0.000 | 166.860 |
| 684.844 | 349.629 | 490.526 | -0.000 | -0.000 | -0.000 |
| 347.910 | 701.366 | 493.026 | 0.000 | 0.000 | 0.000 |
| 494.133 | 495.911 | 1146.733 | -0.000 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.001 | 297.343 | 0.000 | -0.000 |
| 0.001 | 0.000 | -0.001 | -0.000 | 295.949 | -0.000 |
| 0.001 | 0.000 | 0.000 | -0.000 | -0.000 | 166.860 |
| 692.492 | 361.460 | 471.335 | -10.268 | 0.000 | -0.000 |
| 360.923 | 693.063 | 471.354 | 11.976 | 0.000 | 0.000 |
| 467.420 | 467.420 | 939.493 | -0.000 | 0.000 | 0.000 |
| -12.656 | 12.656 | 0.000 | 253.449 | 0.000 | 0.000 |
| 31.125 | 31.125 | 34.986 | 0.000 | 251.344 | -11.973 |
| -0.000 | -0.000 | -0.000 | -0.000 | -12.656 | 166.290 |
| 631.071 | 298.931 | 401.671 | -11.976 | -0.000 | 0.000 |
| 332.689 | 665.270 | 439.866 | 12.656 | -0.000 | 0.000 |
| 439.793 | 439.793 | 908.439 | -0.000 | -0.000 | 0.000 |
| -12.656 | 12.656 | -0.000 | 253.449 | -0.000 | 0.000 |
| -31.125 | -31.125 | -34.986 | -0.000 | 251.344 | -11.973 |
| 0.000 | 0.000 | 0.000 | -0.000 | -12.656 | 166.290 |
| 665.270 | 332.689 | 439.866 | -12.656 | 0.000 | -0.000 |
| 332.689 | 665.270 | 439.866 | 12.656 | 0.000 | -0.000 |
| 439.537 | 439.537 | 908.153 | -0.000 | 0.000 | -0.000 |
| -7.393 | 16.026 | 4.852 | 250.531 | 0.000 | -0.000 |
| 4.316 | 4.316 | 4.852 | 0.000 | 250.531 | -11.710 |
| 3.247 | 3.247 | 3.650 | 0.000 | -11.944 | 166.060 |
| 658.414 | 327.381 | 433.029 | -10.268 | -0.000 | 0.000 |
| 332.689 | 665.270 | 439.865 | 12.656 | -0.000 | 0.000 |
| 433.341 | 433.341 | 901.184 | 0.000 | -0.000 | -0.000 |
| -13.366 | 7.170 | -3.483 | 246.085 | -0.000 | 0.000 |
| -4.316 | -4.316 | -4.852 | -0.000 | 250.531 | -11.710 |
| -3.247 | -3.247 | -3.650 | -0.000 | -11.944 | 166.060 |
| 662.188 | 330.078 | 436.667 | -11.933 | 0.000 | -0.000 |
| 332.690 | 665.268 | 439.866 | 12.655 | 0.000 | -0.000 |
| 436.750 | 436.750 | 905.035 | -0.000 | 0.000 | -0.000 |
| -9.307 | 11.404 | 1.180 | 246.355 | 0.000 | -0.000 |
| 0.309 | 0.310 | 0.349 | 0.000 | 246.085 | -10.268 |
| 0.309 | 0.310 | 0.348 | 0.000 | -10.268 | 165.516 |
| 665.272 | 332.690 | 439.865 | -12.656 | -0.000 | -0.000 |
| 329.484 | 661.609 | 436.005 | 11.948 | -0.000 | 0.000 |
| 439.792 | 439.792 | 908.421 | 0.000 | -0.000 | 0.000 |
| -10.578 | 9.959 | -0.349 | 246.086 | 0.000 | -0.000 |
| -0.309 | -0.310 | -0.349 | -0.000 | 246.085 | -10.268 |
| -0.309 | -0.310 | -0.348 | -0.000 | -10.268 | 165.516 |
| 665.953 | 332.608 | 439.017 | -12.823 | -0.000 | 0.000 |
| 332.608 | 665.953 | 439.017 | 12.823 | -0.000 | 0.000 |
| 439.017 | 439.017 | 900.185 | -0.000 | 0.000 | -0.000 |
| -10.460 | 10.529 | 0.039 | 245.843 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.000 | 0.000 | 253.134 | -12.823 |
| 0.035 | 0.035 | 0.039 | 0.000 | -10.496 | 165.929 |
| 662.610 | 329.711 | 435.507 | -12.126 | 0.000 | -0.000 |
| 329.711 | 662.610 | 435.507 | 12.126 | 0.000 | -0.000 |
| 439.017 | 439.017 | 900.185 | 0.000 | 0.000 | 0.000 |
| -12.823 | 12.823 | -0.000 | 253.134 | 0.000 | 0.000 |
| 0.000 | 0.000 | -0.000 | -0.000 | 253.134 | -12.823 |
| -0.035 | -0.035 | -0.039 | -0.000 | -10.496 | 165.929 |
| 665.953 | 332.608 | 439.017 | -12.823 | -0.000 | -0.000 |
| 332.609 | 665.953 | 439.017 | 12.823 | 0.000 | -0.000 |
| 439.017 | 439.017 | 900.187 | -0.000 | -0.000 | 0.000 |
| -12.823 | 12.823 | 0.000 | 253.134 | -0.000 | -0.000 |
| 0.003 | 0.003 | 0.004 | 0.000 | 245.843 | -10.495 |
| -0.000 | -0.000 | -0.000 | 0.000 | -12.823 | 166.672 |
| 665.953 | 332.609 | 439.017 | -12.823 | 0.000 | -0.000 |
| 332.608 | 665.953 | 439.017 | 12.823 | 0.000 | -0.000 |
| 435.684 | 435.684 | 896.435 | 0.000 | 0.000 | -0.000 |
| -12.823 | 12.823 | -0.000 | 253.134 | -0.000 | 0.000 |
| -0.003 | -0.003 | -0.004 | -0.000 | 245.843 | -10.495 |
| 0.000 | 0.000 | 0.000 | -0.000 | -12.823 | 166.672 |
| 665.951 | 332.608 | 439.017 | -12.823 | 0.000 | -0.000 |
| 332.609 | 665.951 | 439.017 | 12.823 | -0.000 | 0.000 |
| 435.687 | 435.687 | 896.456 | -0.000 | -0.000 | -0.000 |
| -12.823 | 12.822 | 0.001 | 253.134 | -0.000 | -0.000 |
| -0.000 | 0.000 | 0.001 | 0.000 | 245.843 | -10.495 |
| -0.001 | -0.000 | -0.000 | 0.000 | -12.823 | 166.672 |
| 662.249 | 330.390 | 435.685 | -10.495 | 0.000 | 0.000 |
| 332.608 | 665.955 | 439.016 | 12.823 | 0.000 | 0.000 |
| 435.683 | 435.683 | 896.420 | 0.000 | 0.000 | 0.000 |
| -12.823 | 12.823 | -0.001 | 253.134 | -0.000 | -0.000 |
| 0.000 | -0.000 | -0.001 | -0.000 | 245.843 | -10.495 |
| 0.001 | 0.000 | 0.000 | -0.000 | -12.823 | 166.672 |
Phonon and Quasi-Harmonic Approximation Predictions
Phonon band structures and crystal properties estimated from quasi-harmonic approximation (QHA) calculations are displayed for select crystals. The calculations were performed using phonopy and LAMMPS. For the phonon calculations, 3x3 supercells of the potential-specific relaxed crystals were used. The QHA calculations were based on 11 strain states ranging from -0.05 to 0.05.
The calculation method used is available as the iprPy phonon calculation method.
Notes and Disclaimers:
- The thermodynamic properties estimated from QHA are based on the assumption that only volumetric changes affect the phonon behaviors as the temperature changes. This tends to give good predictions at lower temperatures but ignores anharmonic effects such as phonon coupling and vacancy formation that can be important at higher temperatures.
- Note that direct molecular dynamics (MD) simulations using the same potentials will disagree with the thermodynamics properties listed here for the lowest temperatures. The MD results are purely classical in nature and therefore lack a zero-point energy, whereas the phonon calculations inherently provide a zero-point energy.
- The structures explored here are taken from the dynamically relaxed structures above. Despite the rigorous relaxation method used, some of these structures prove to be unstable once internal deformations are added. The phonon results may reflect this and give bad band gap predictions for these unstable crystals.
- All QHA calculations performed here use the same set of strains which might not be ideal for all crystals. Be sure to check the bulk modulus and Helmholtz vs. volume plots to verify if the QHA strained states are reasonable for each crystal of interest.
- QHA results may not be available for all crystal structures that have phonon results. Missing QHA results indicates an issue either with the strained states or with the QHA calculation itself.
- NIST disclaimer
Version Information:
- 2023-03-14. Phonon and QHA plots added
Band Structures, Density of States, and QHA Verification Plots
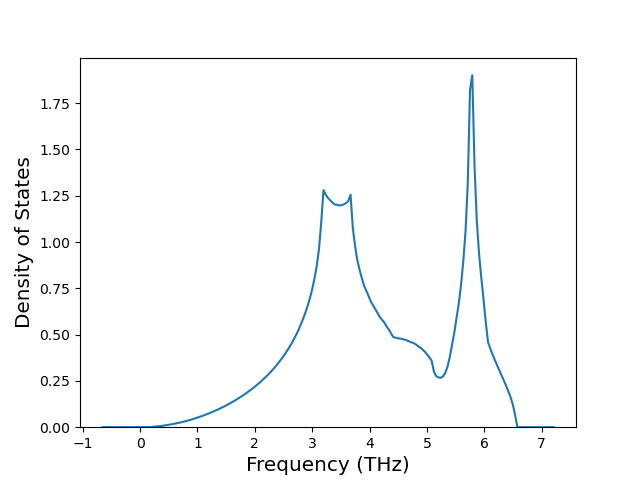
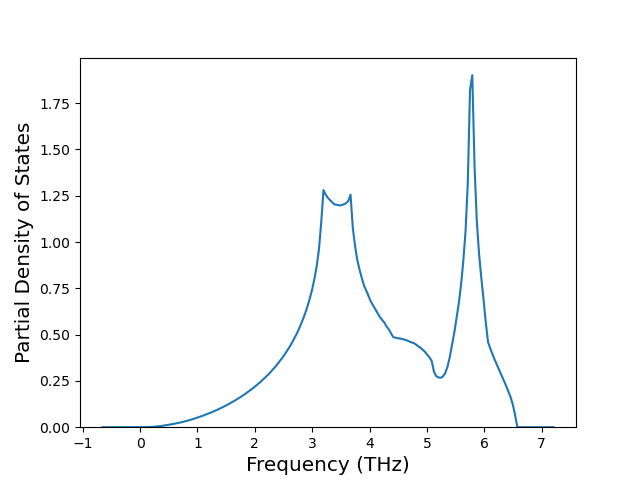
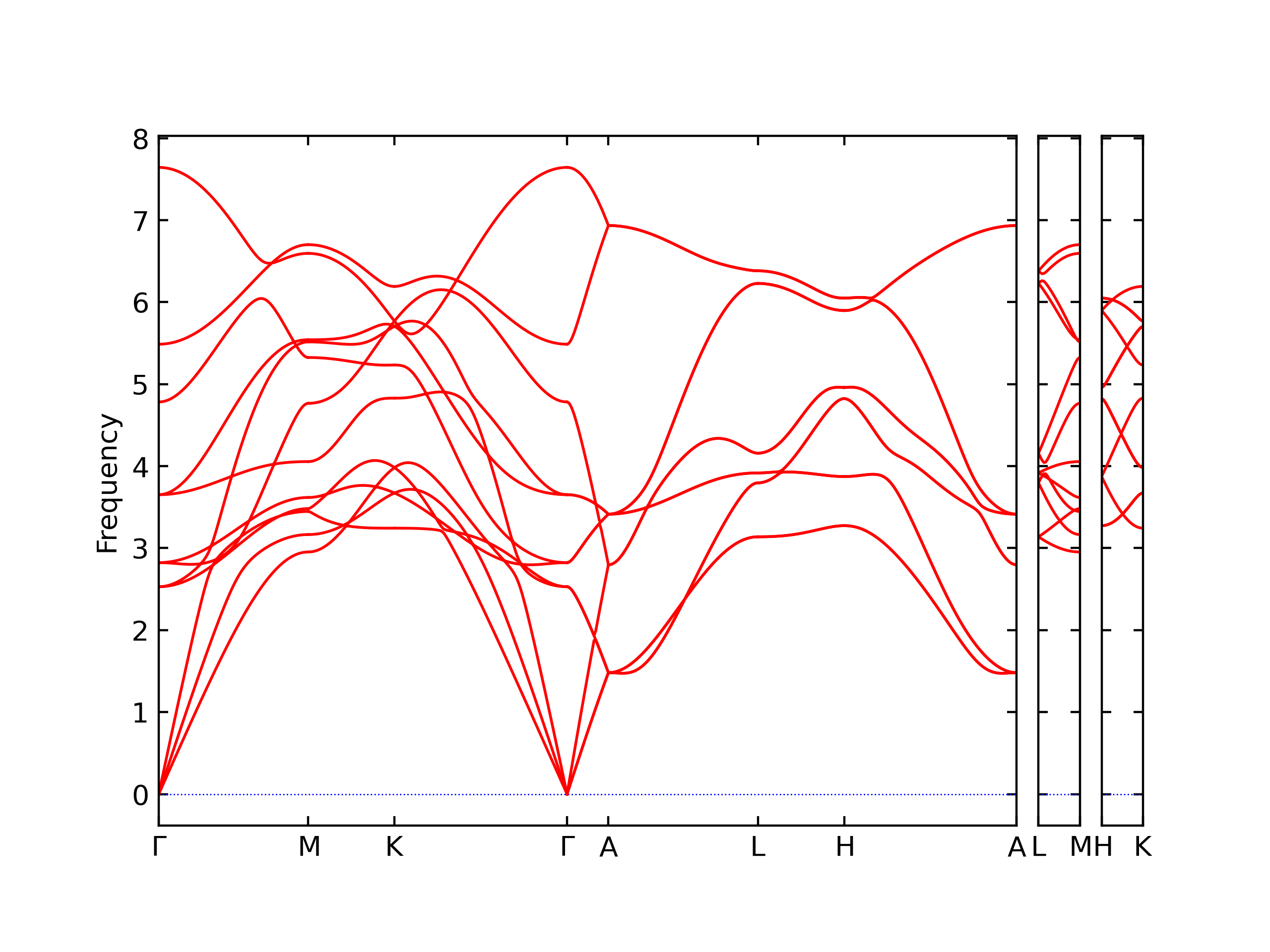
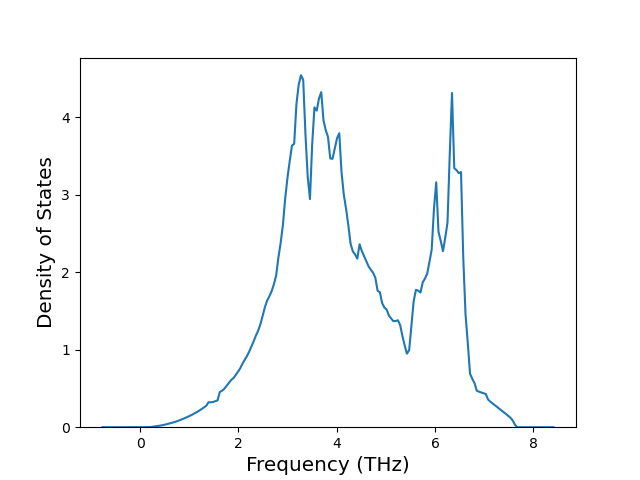
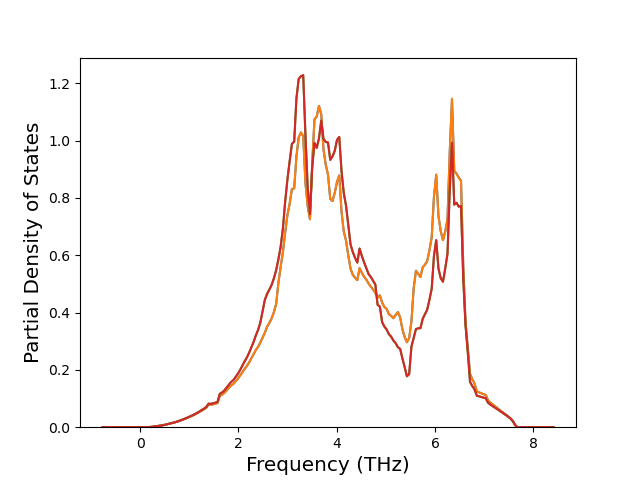
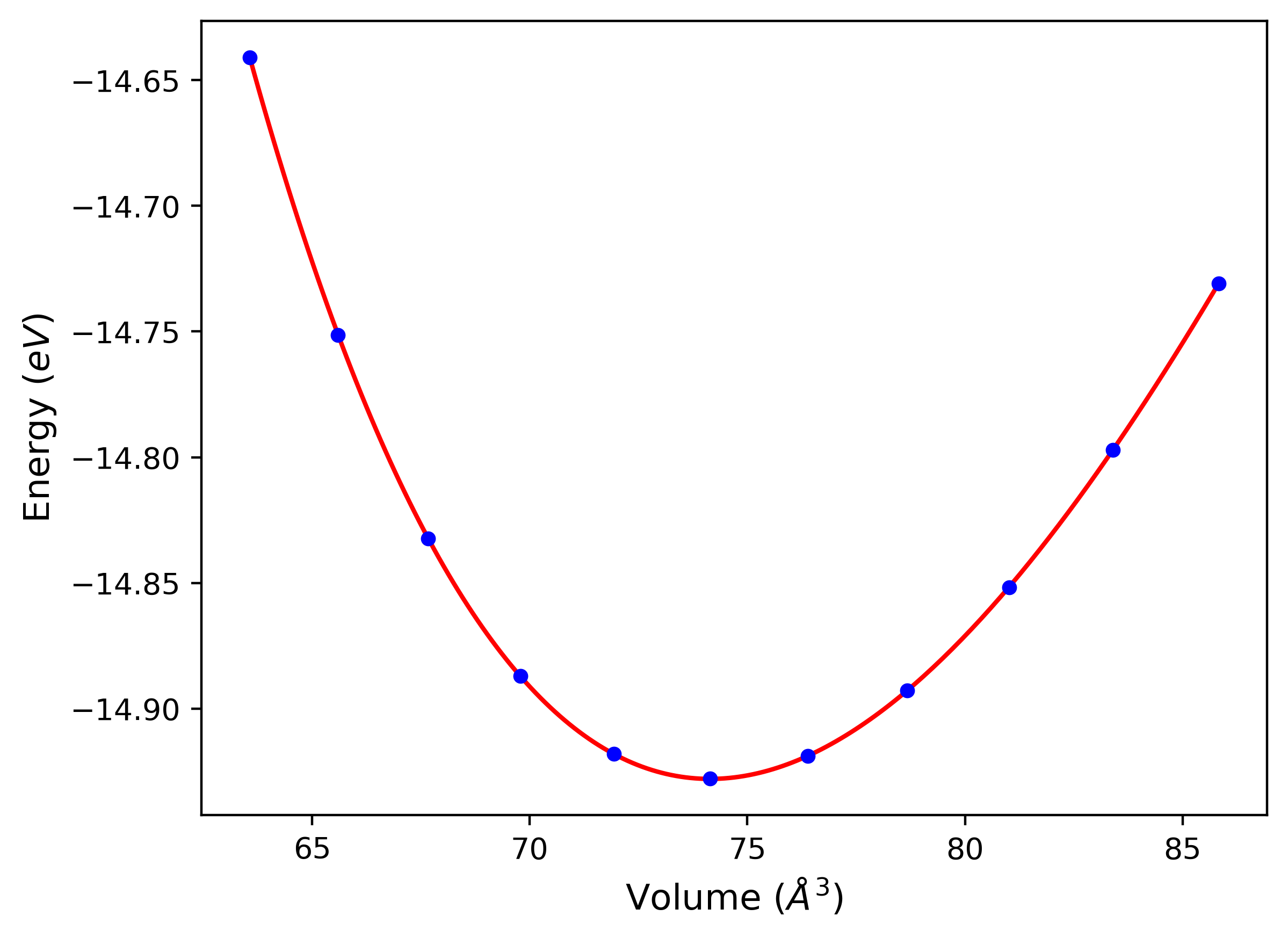
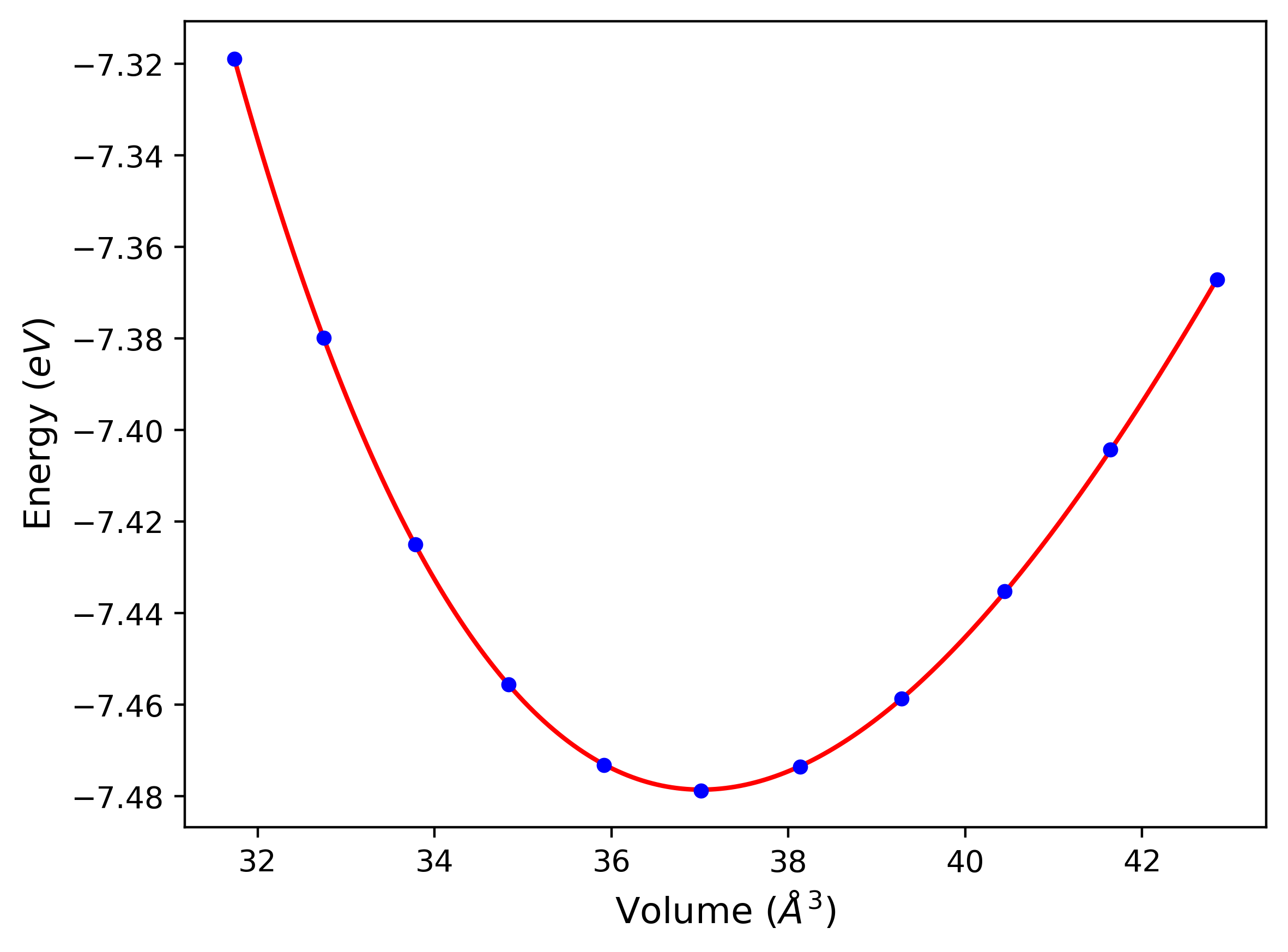
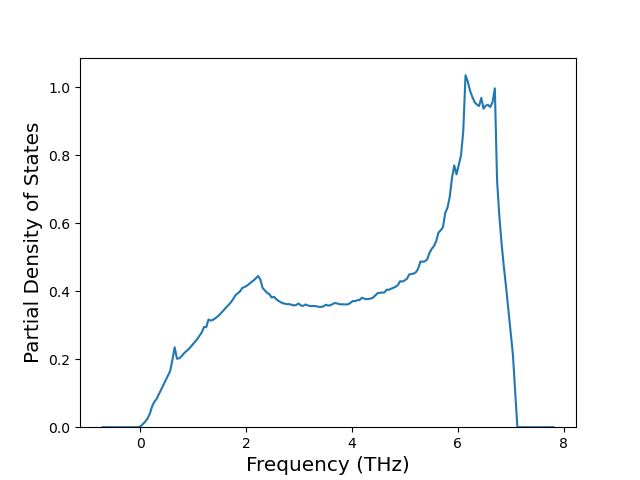
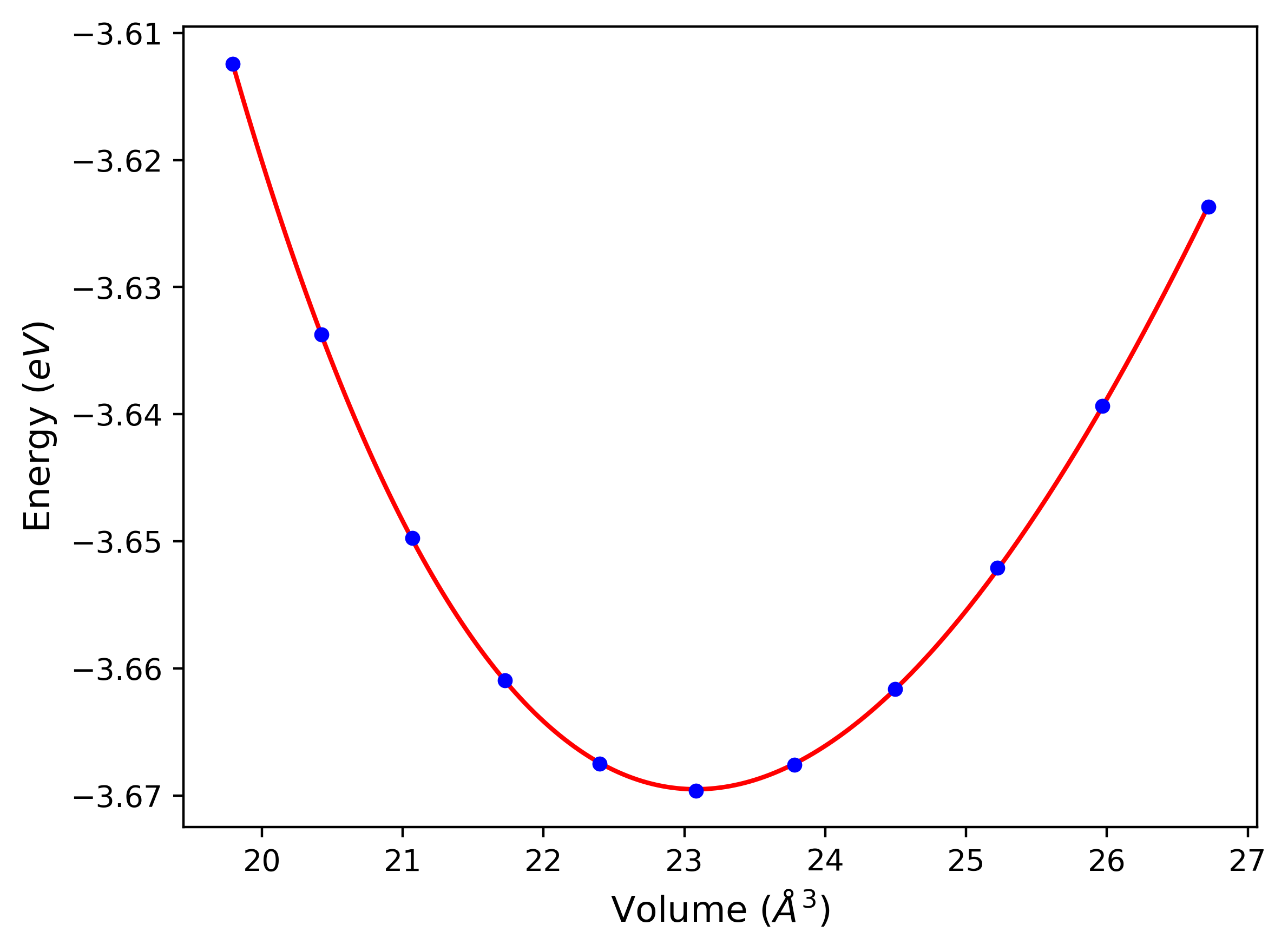
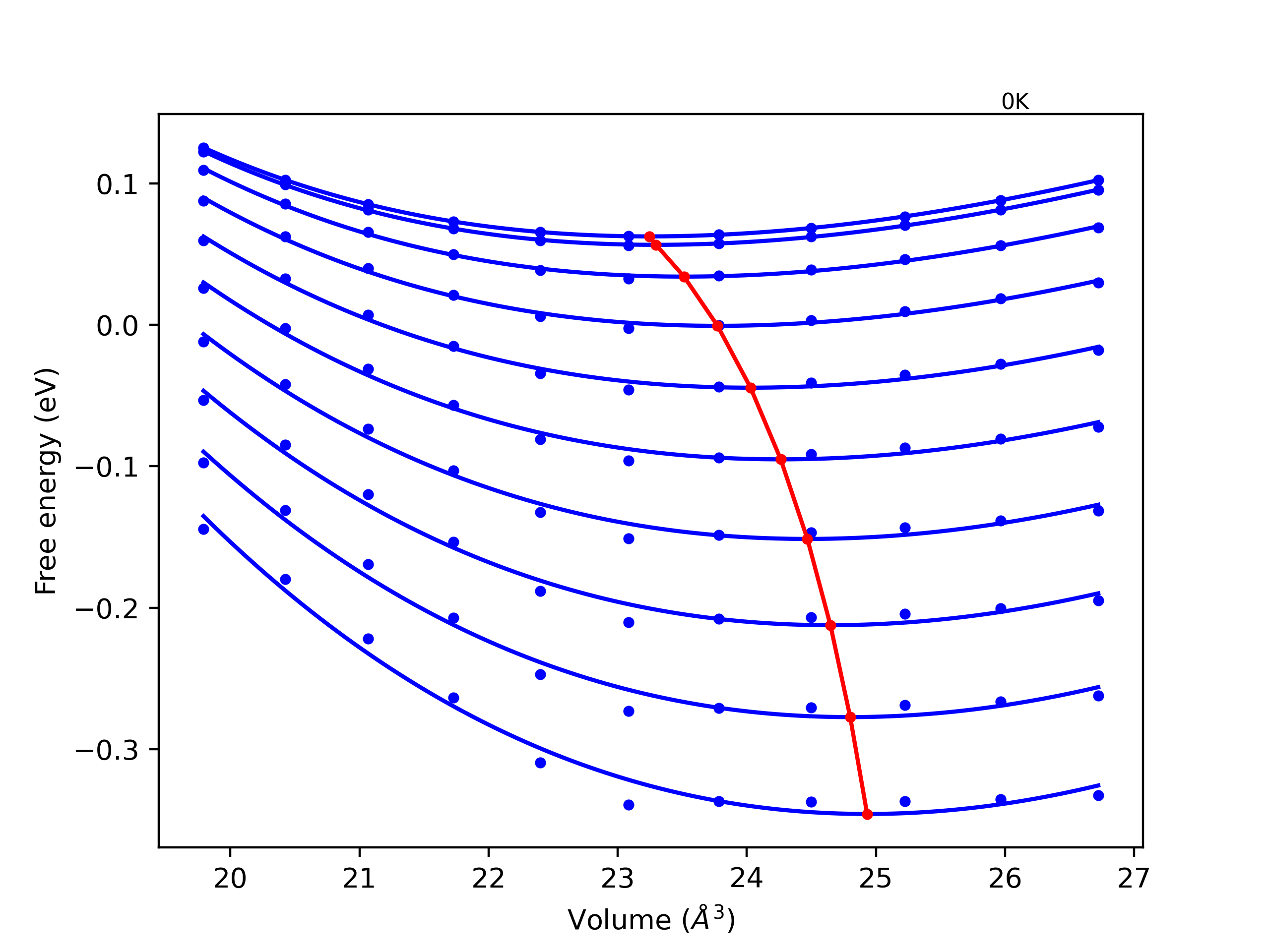
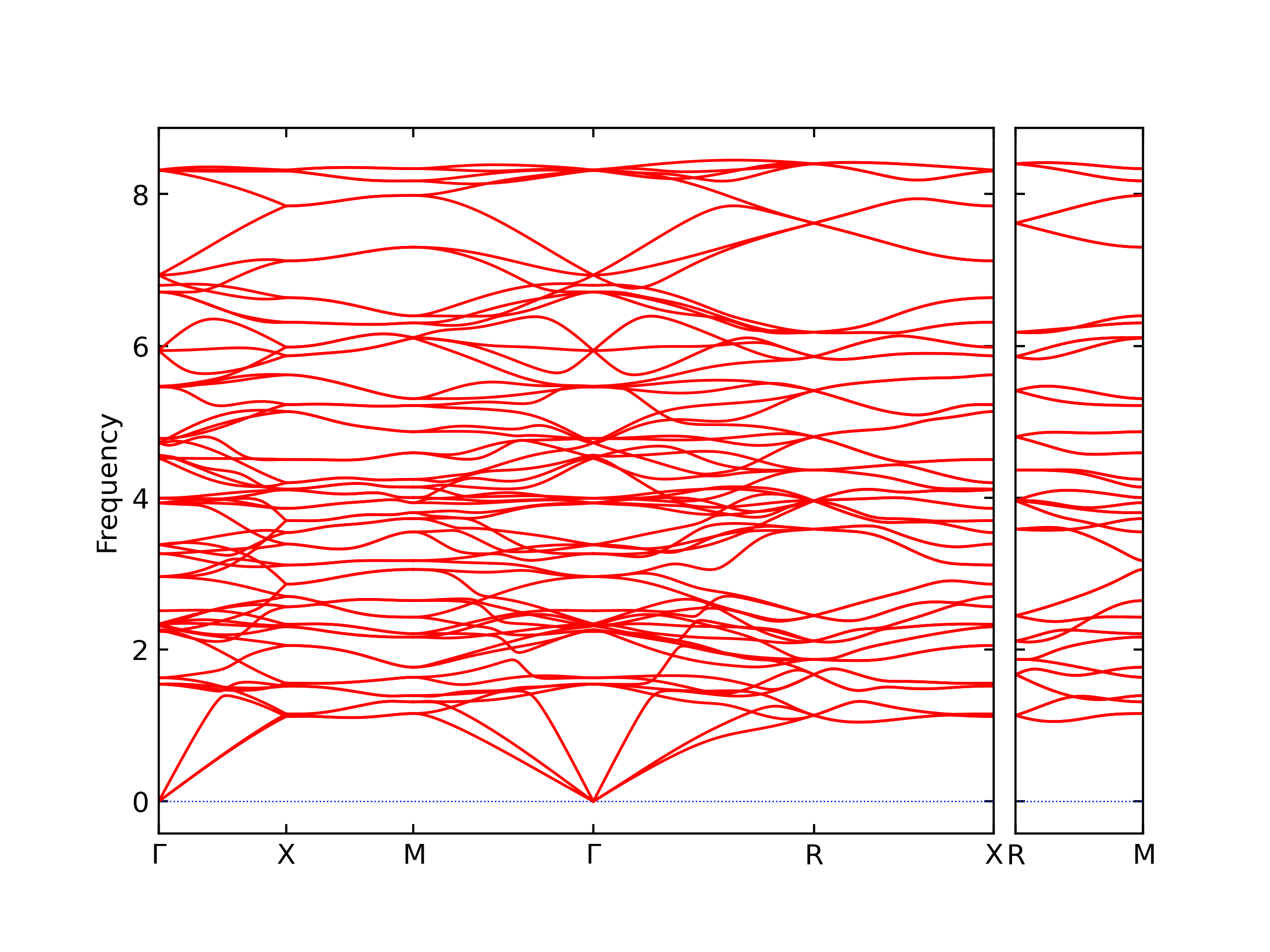
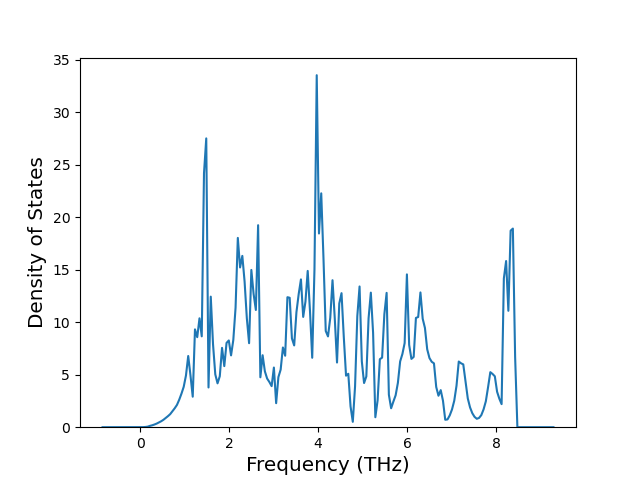
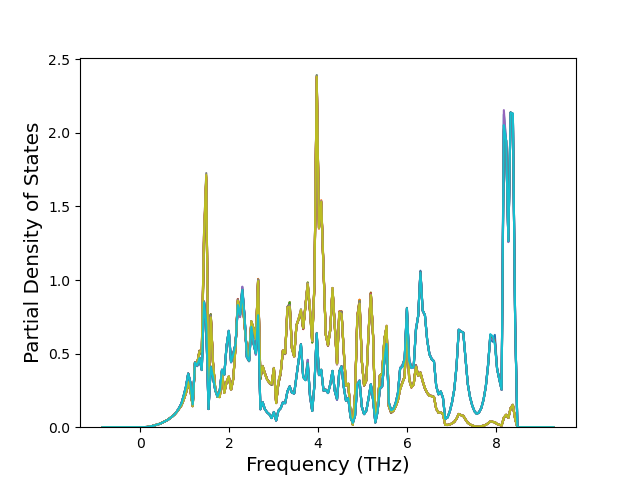
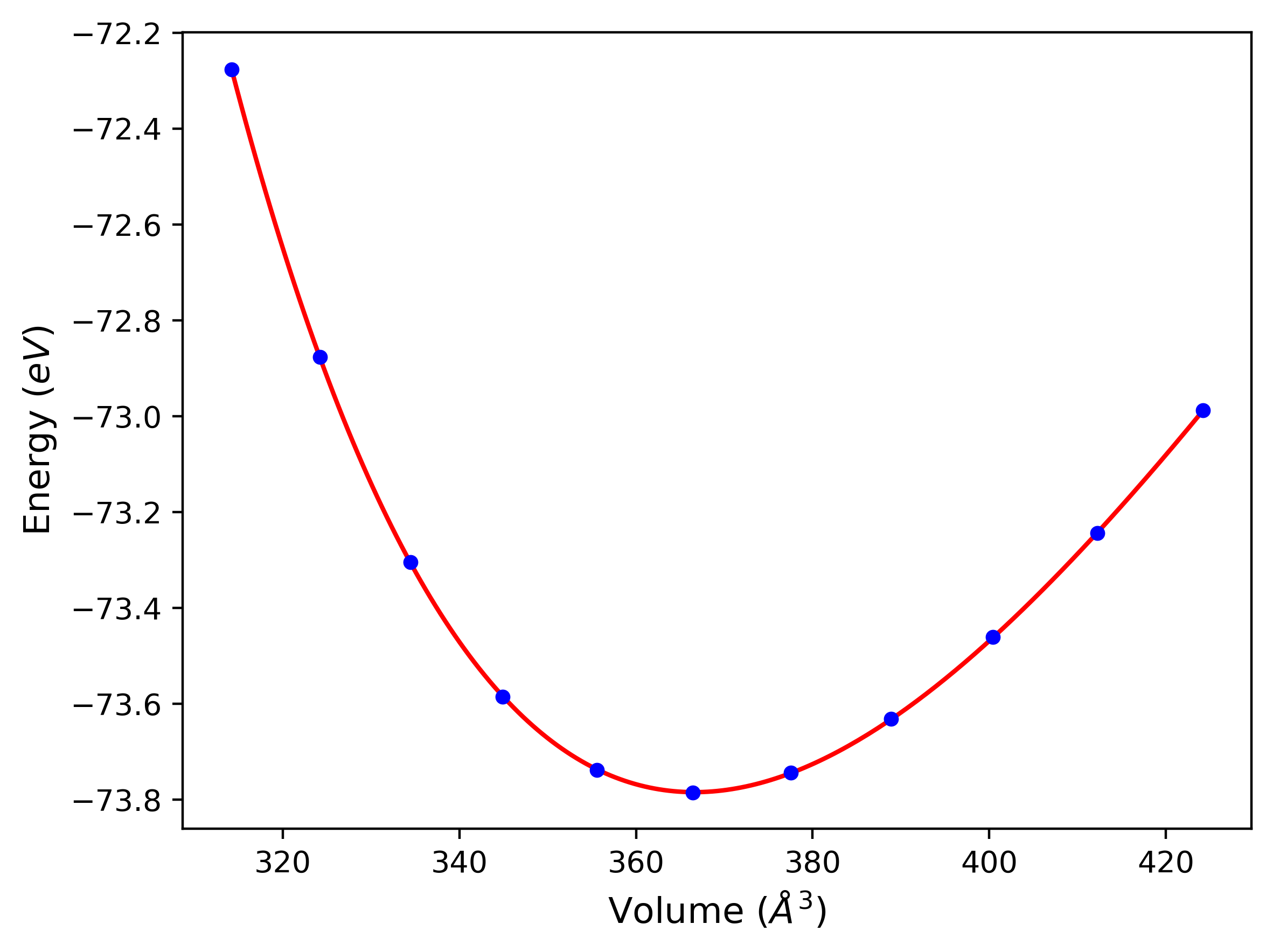
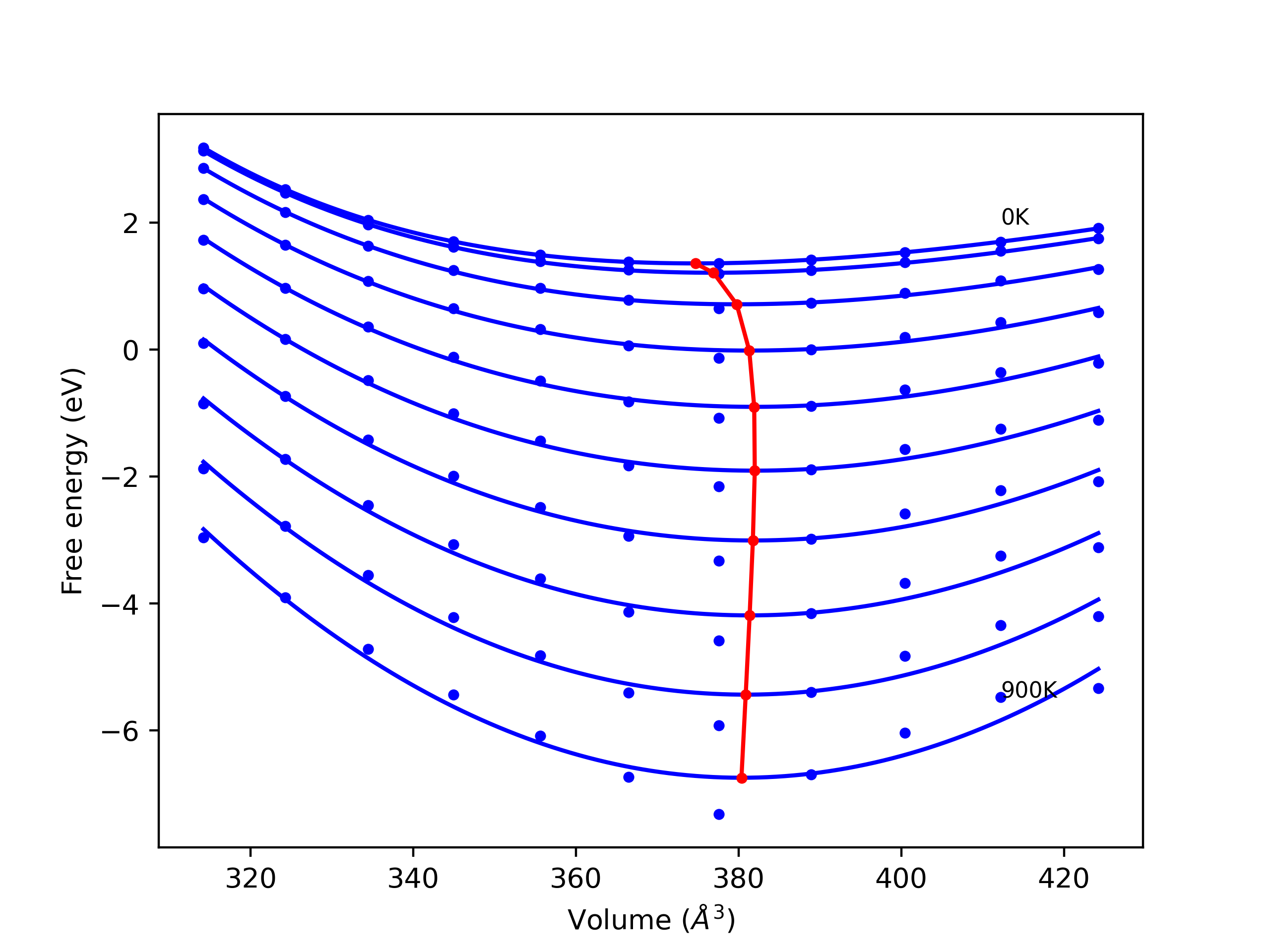
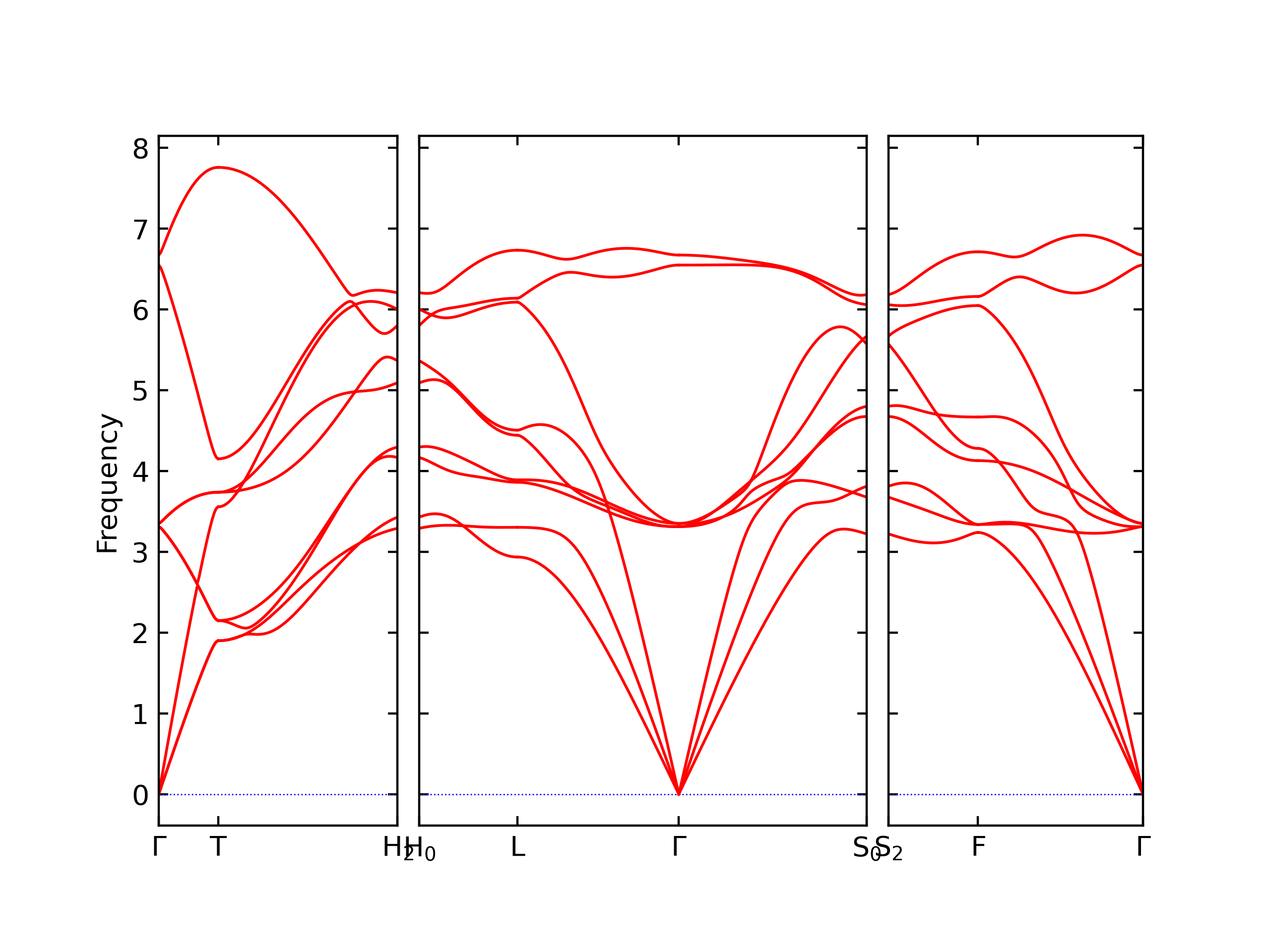
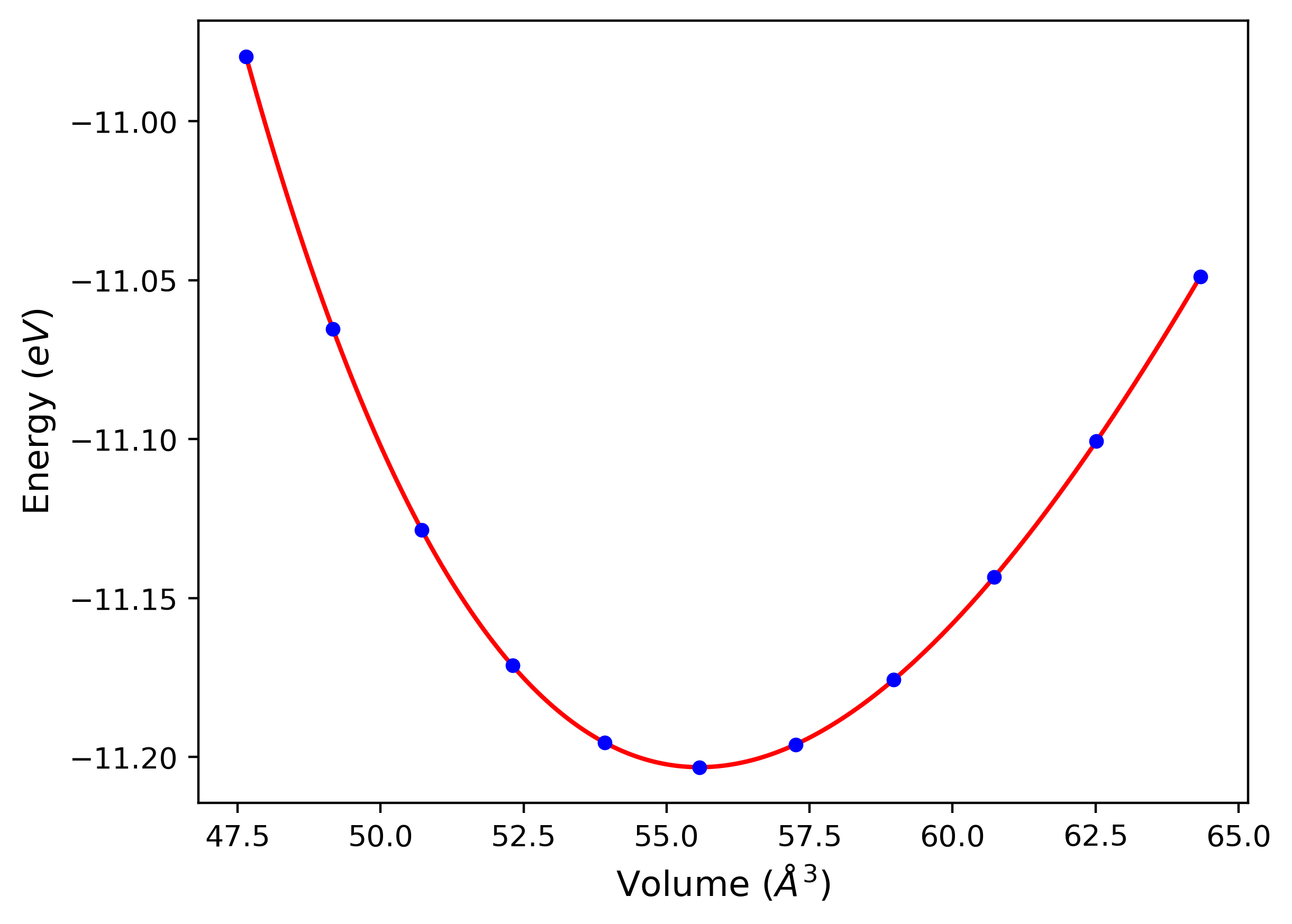
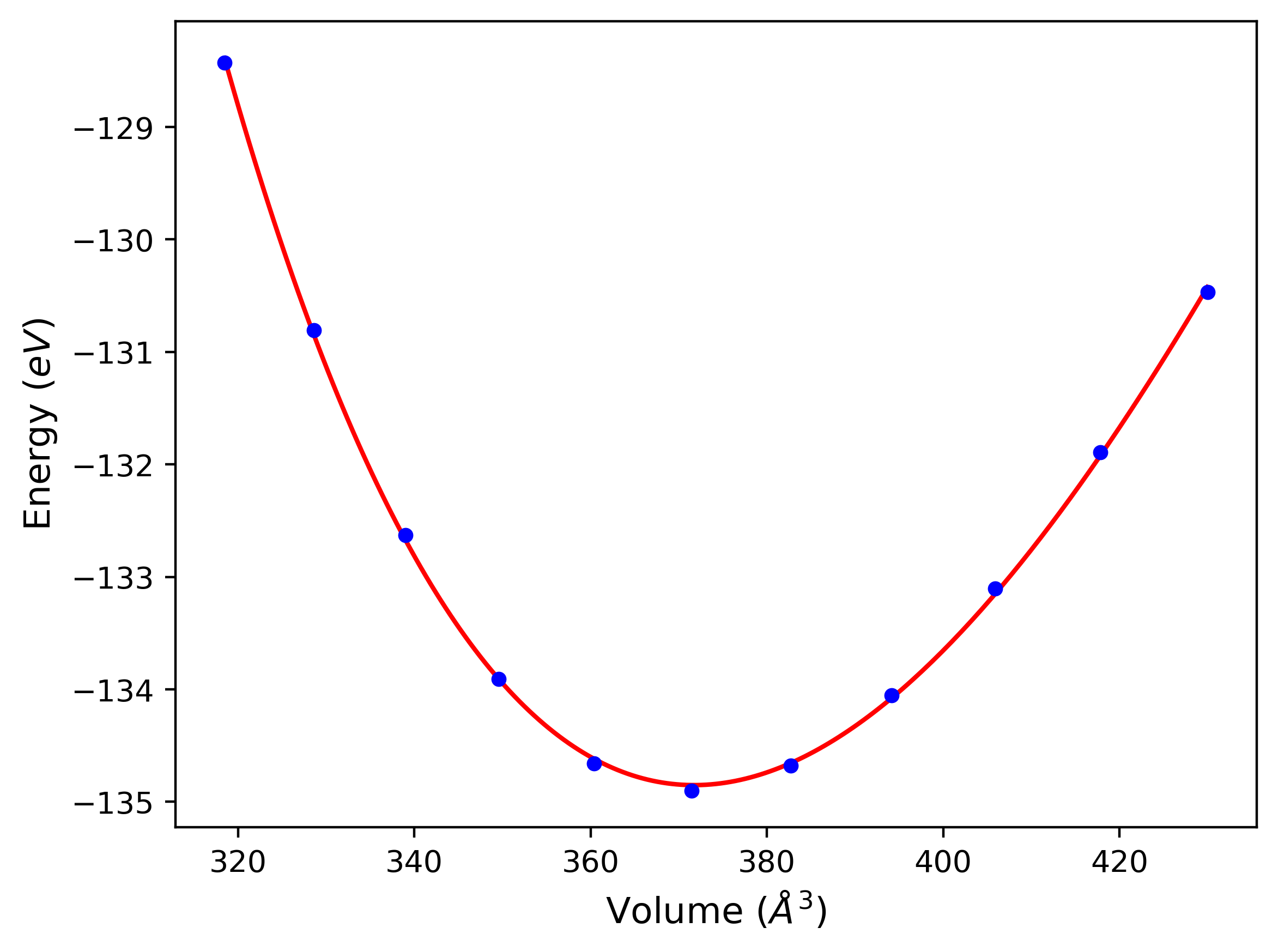
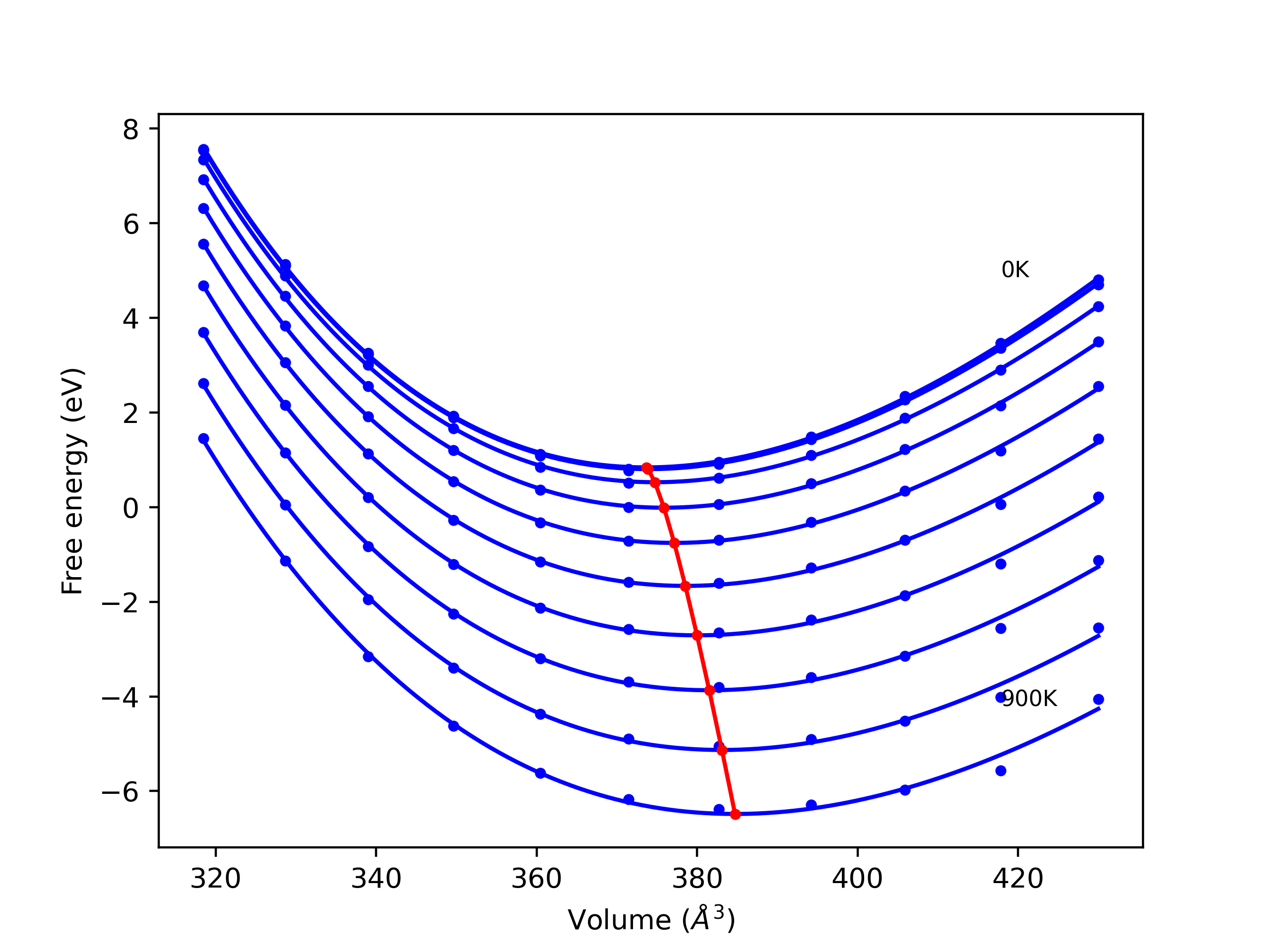
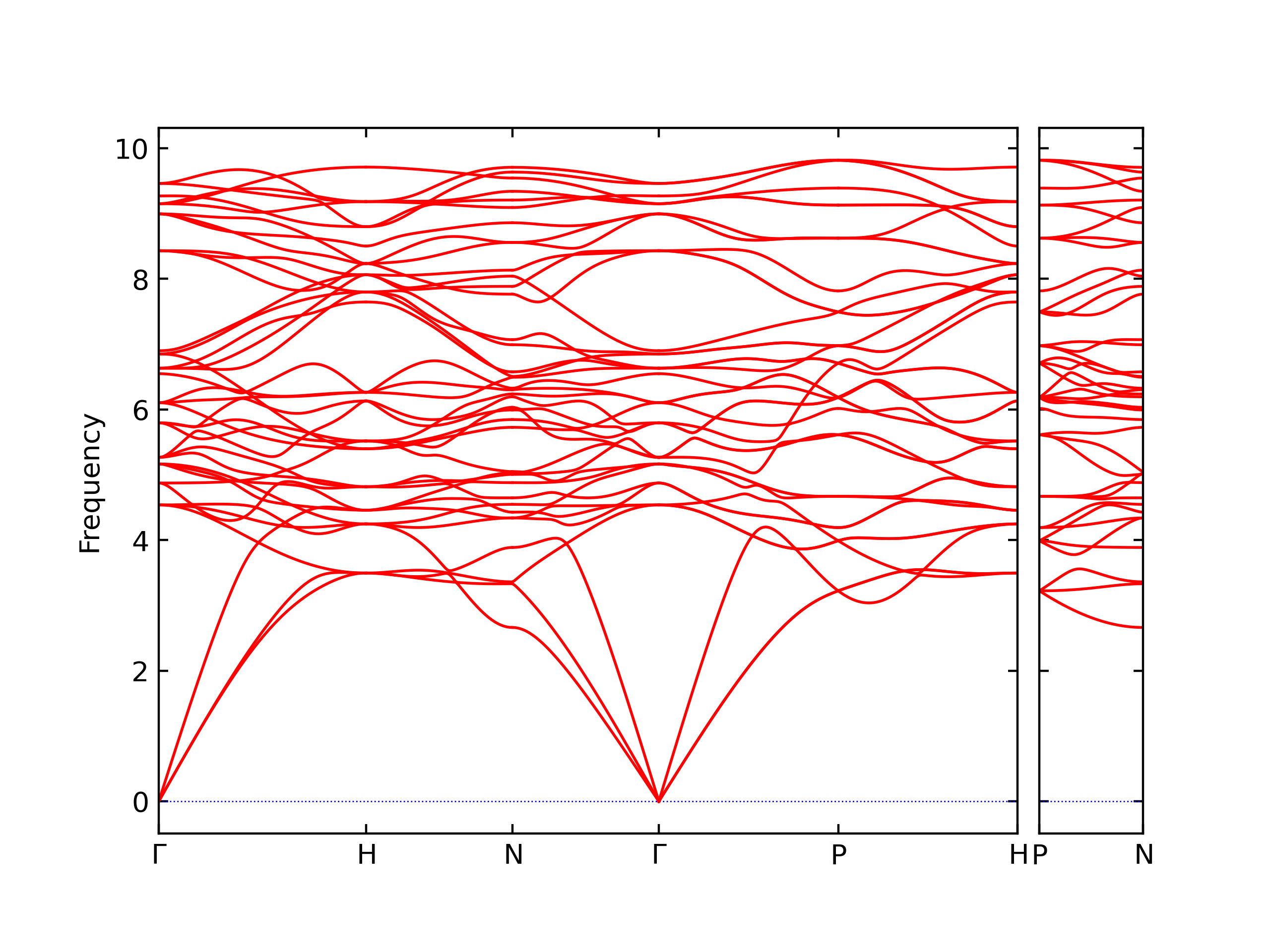
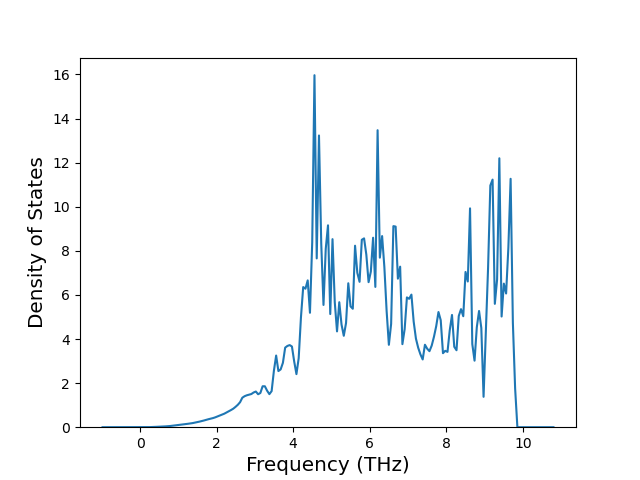
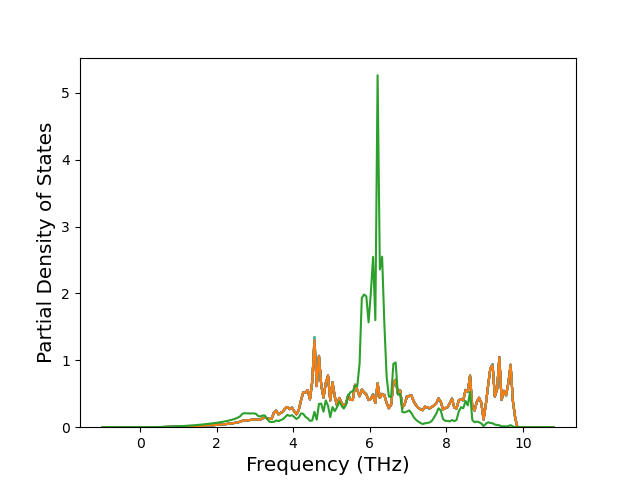
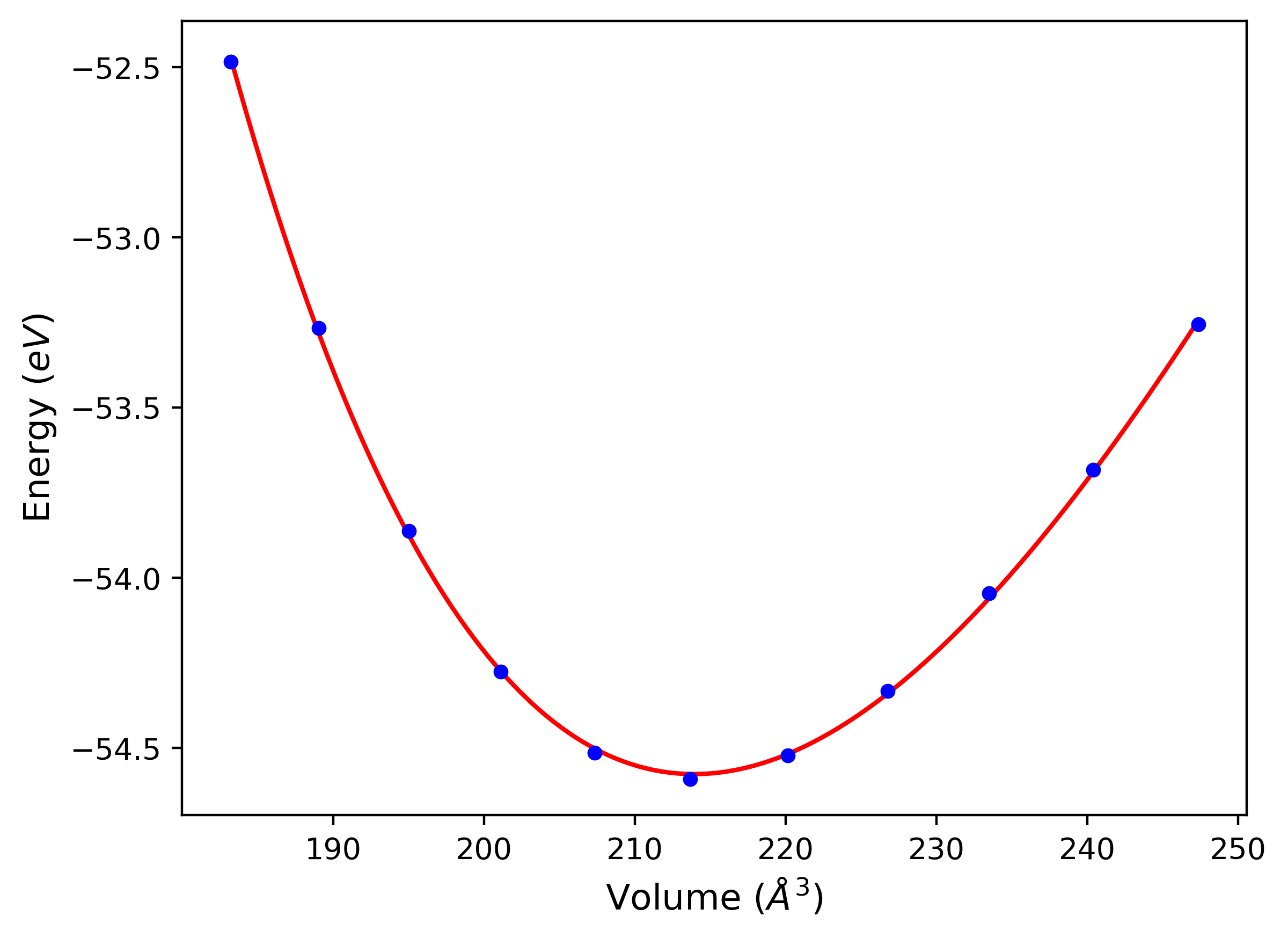
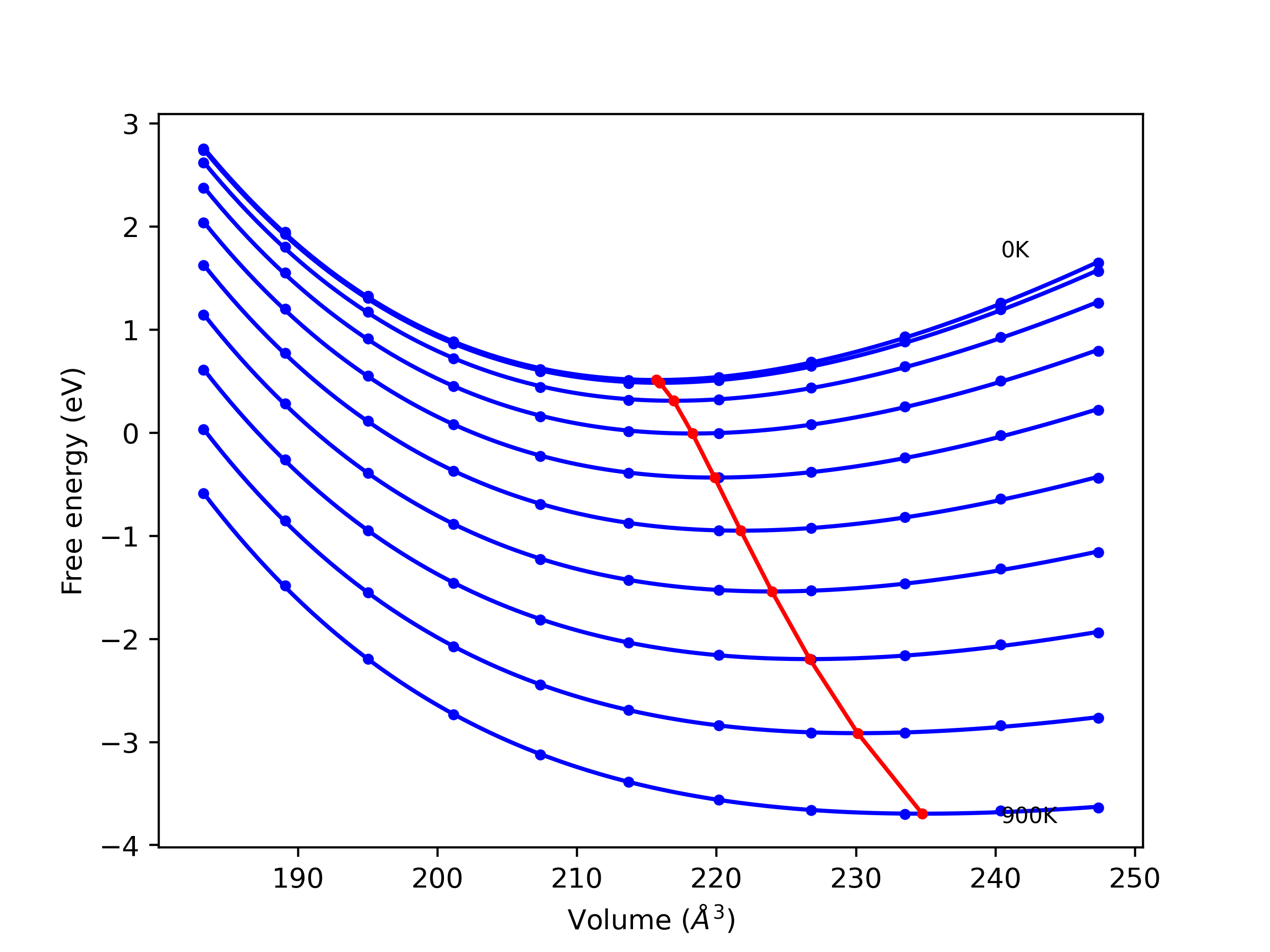
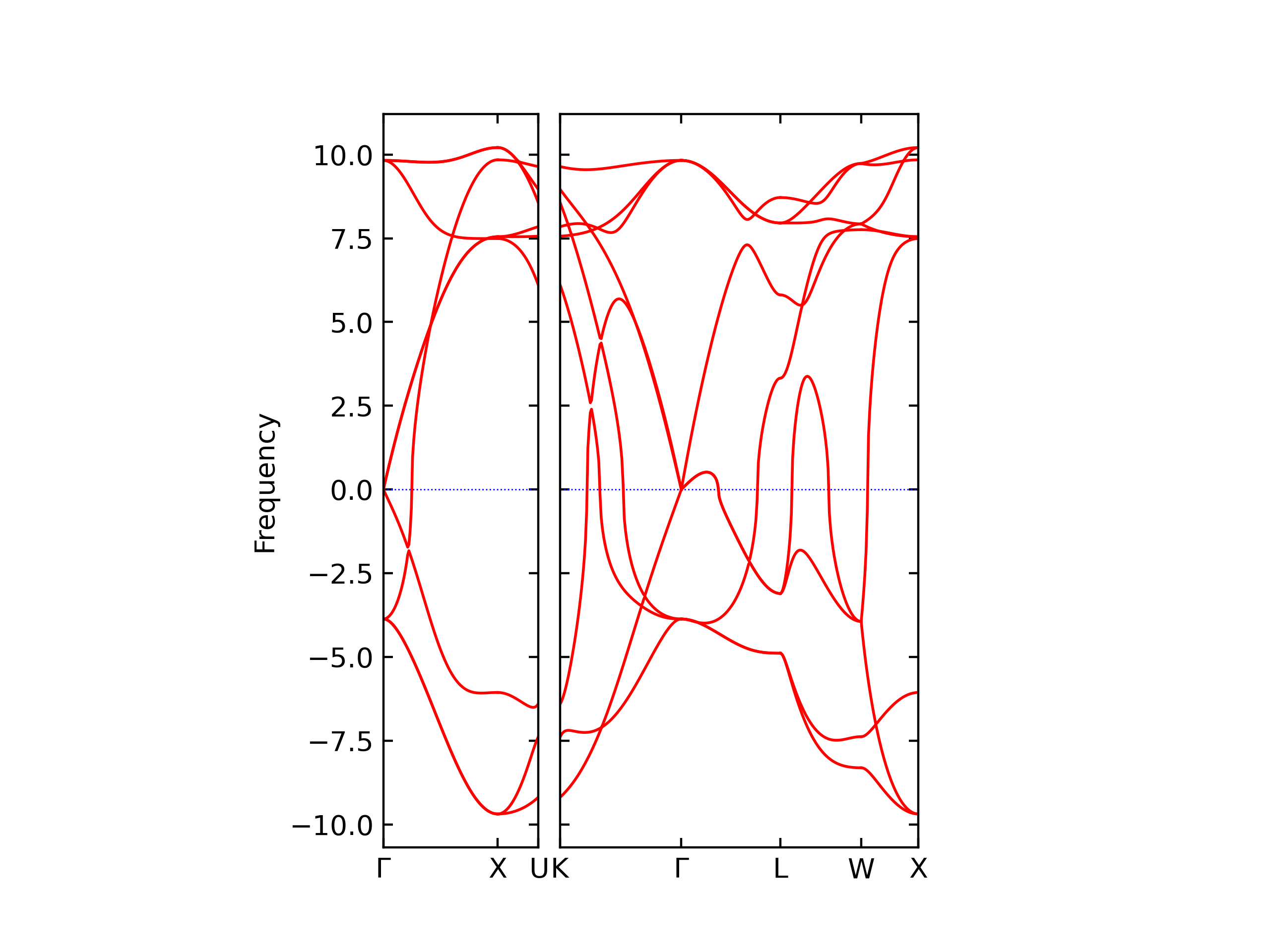
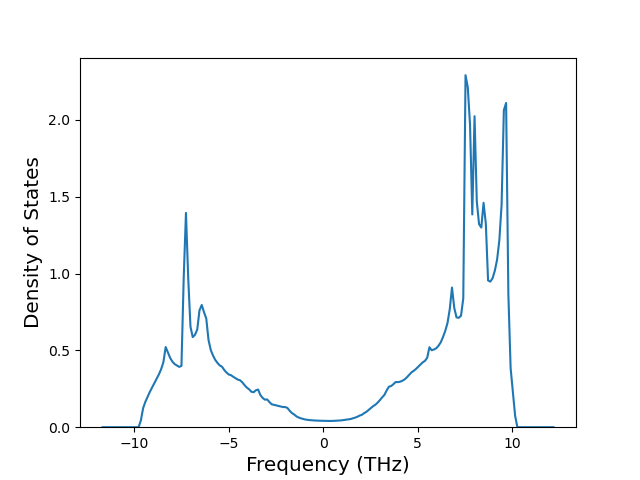
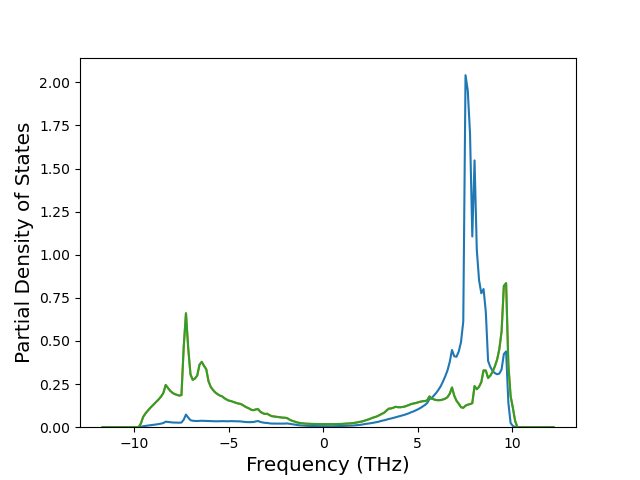
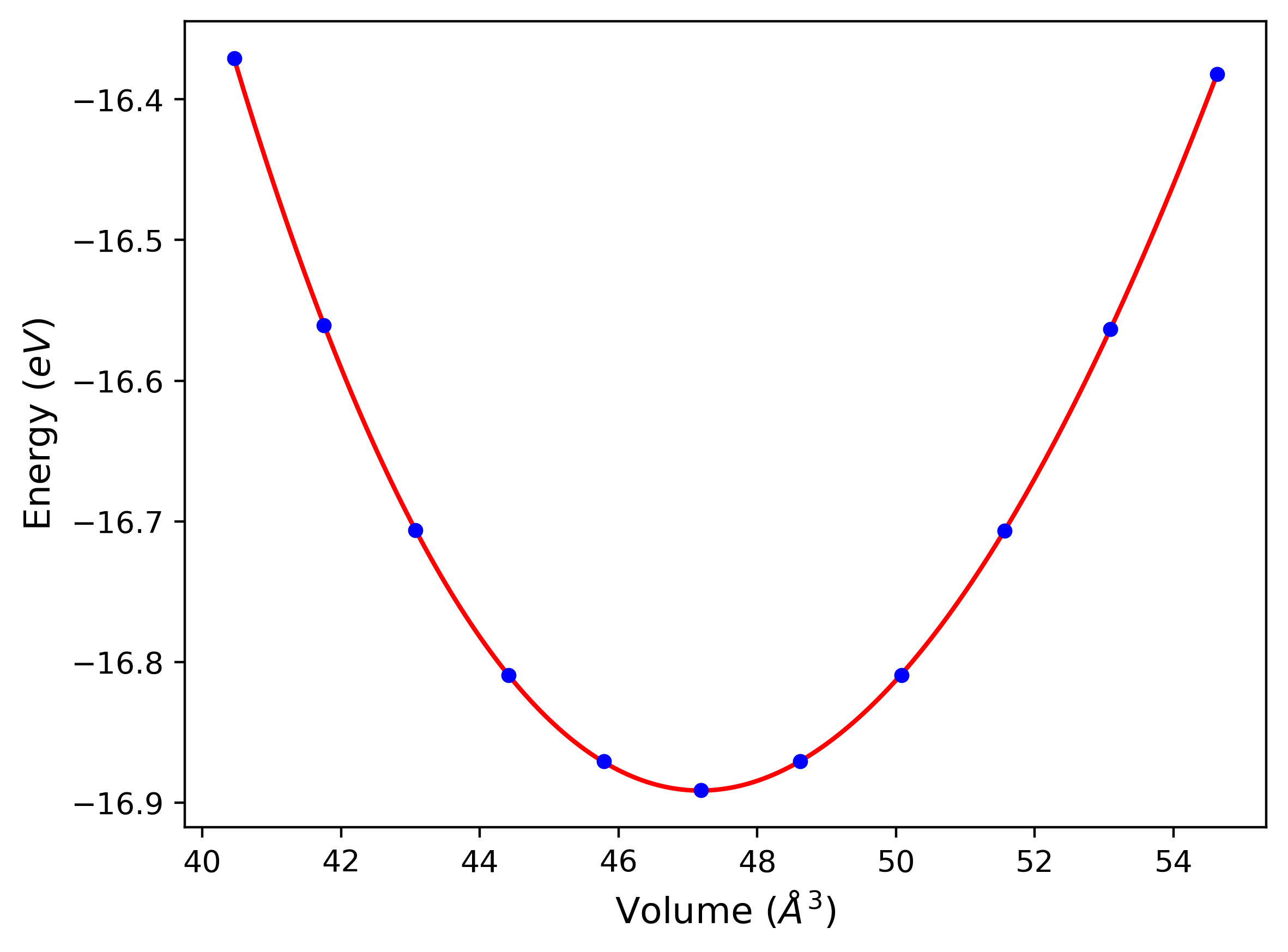
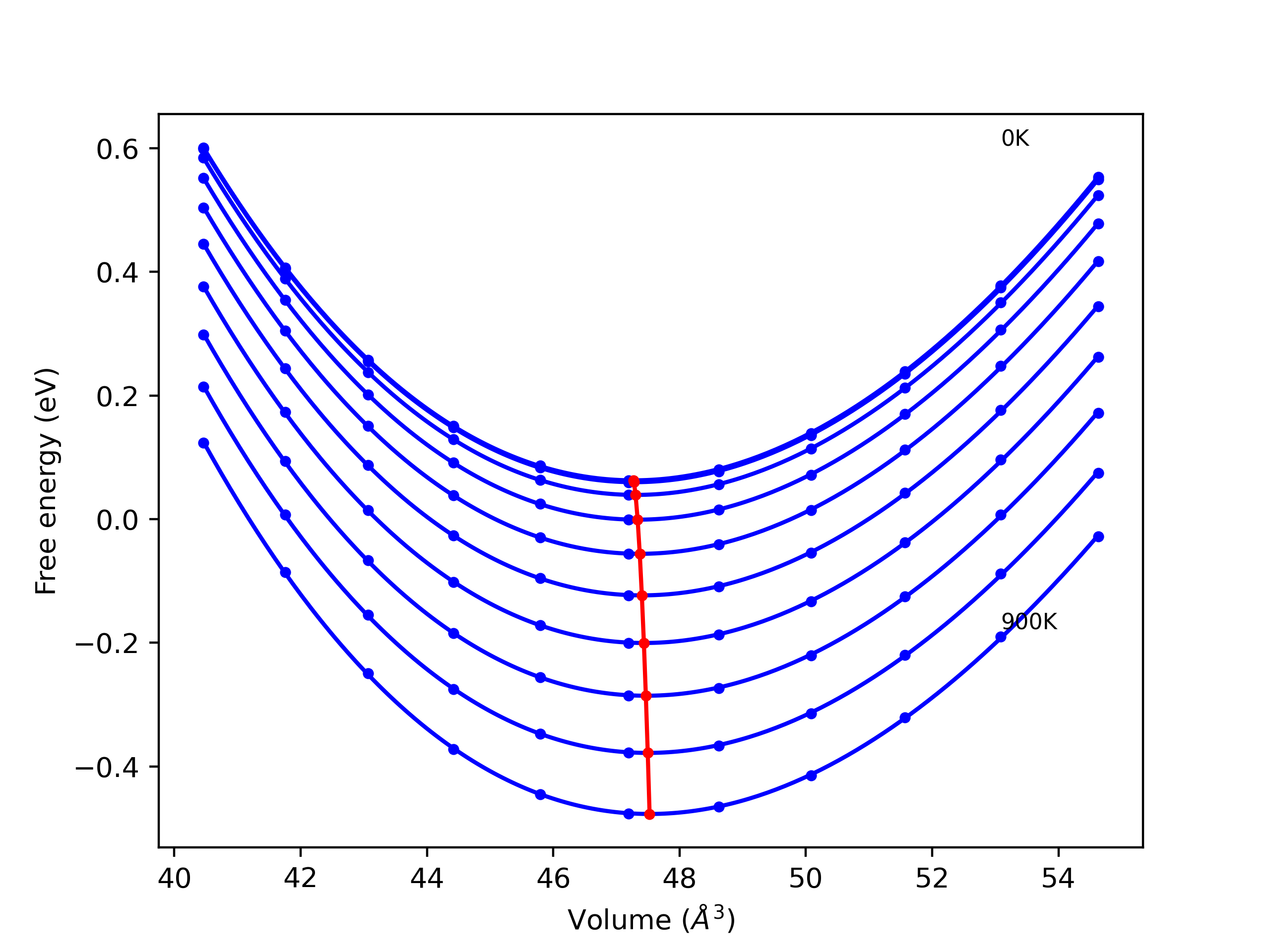
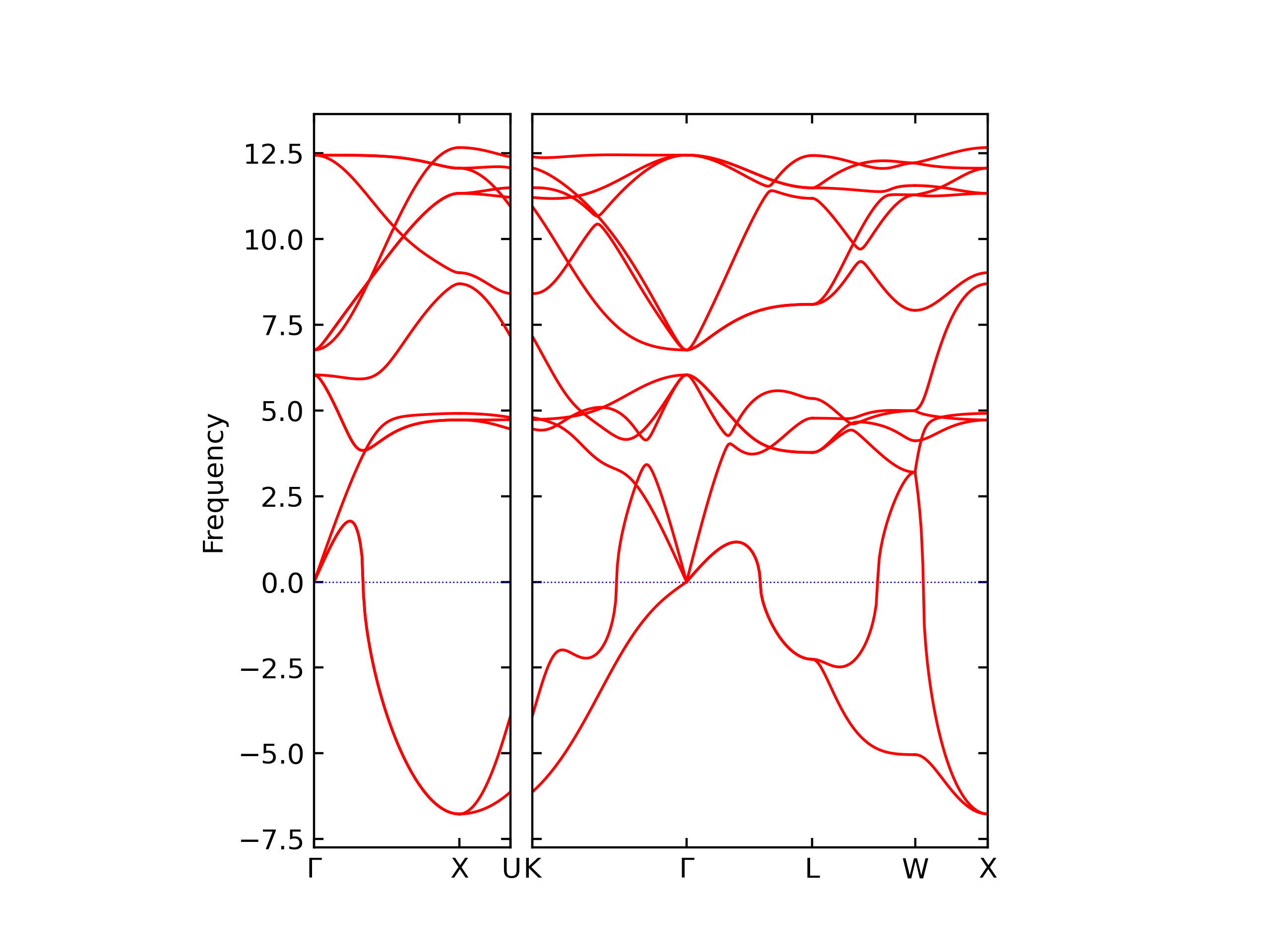
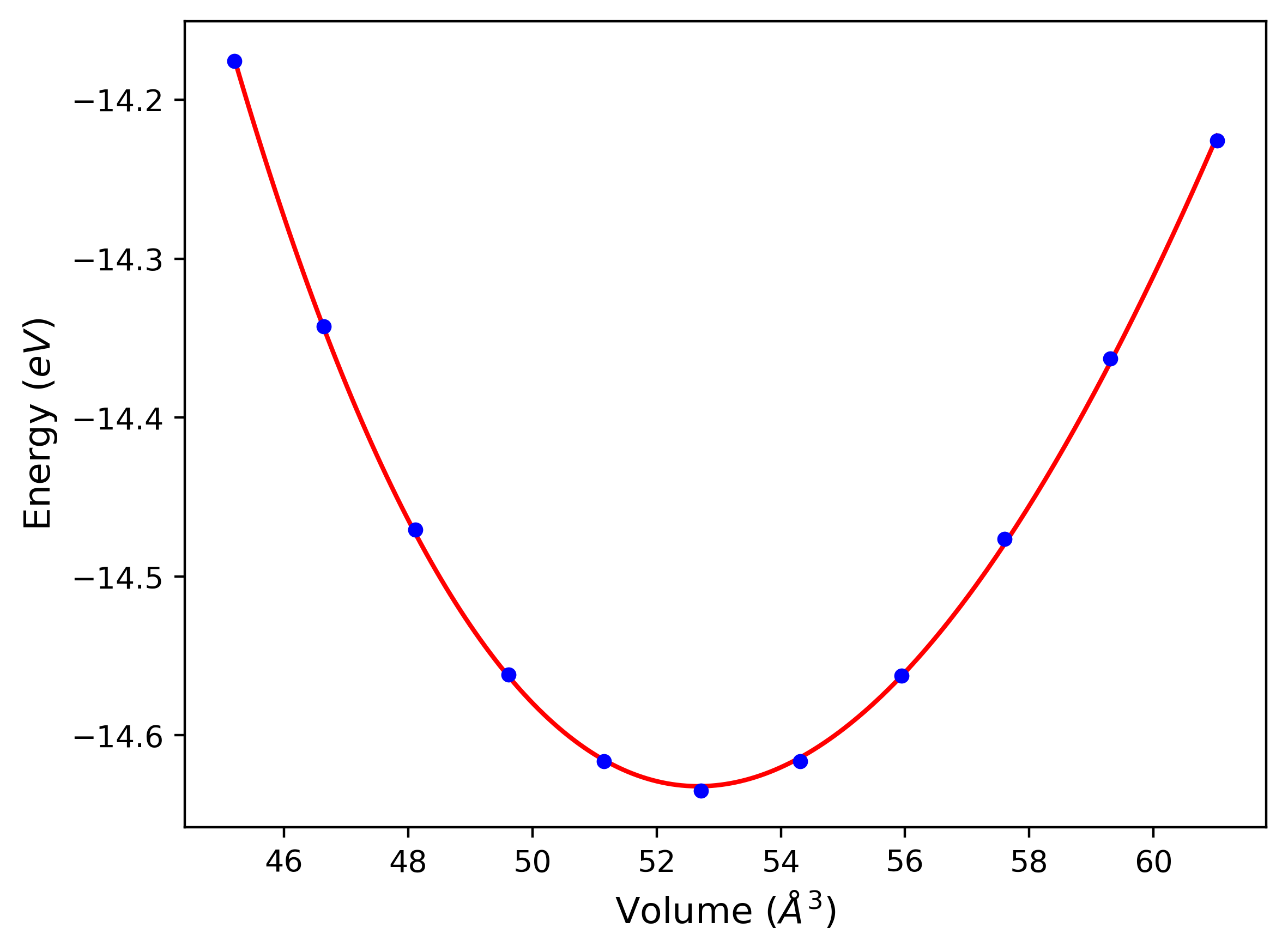
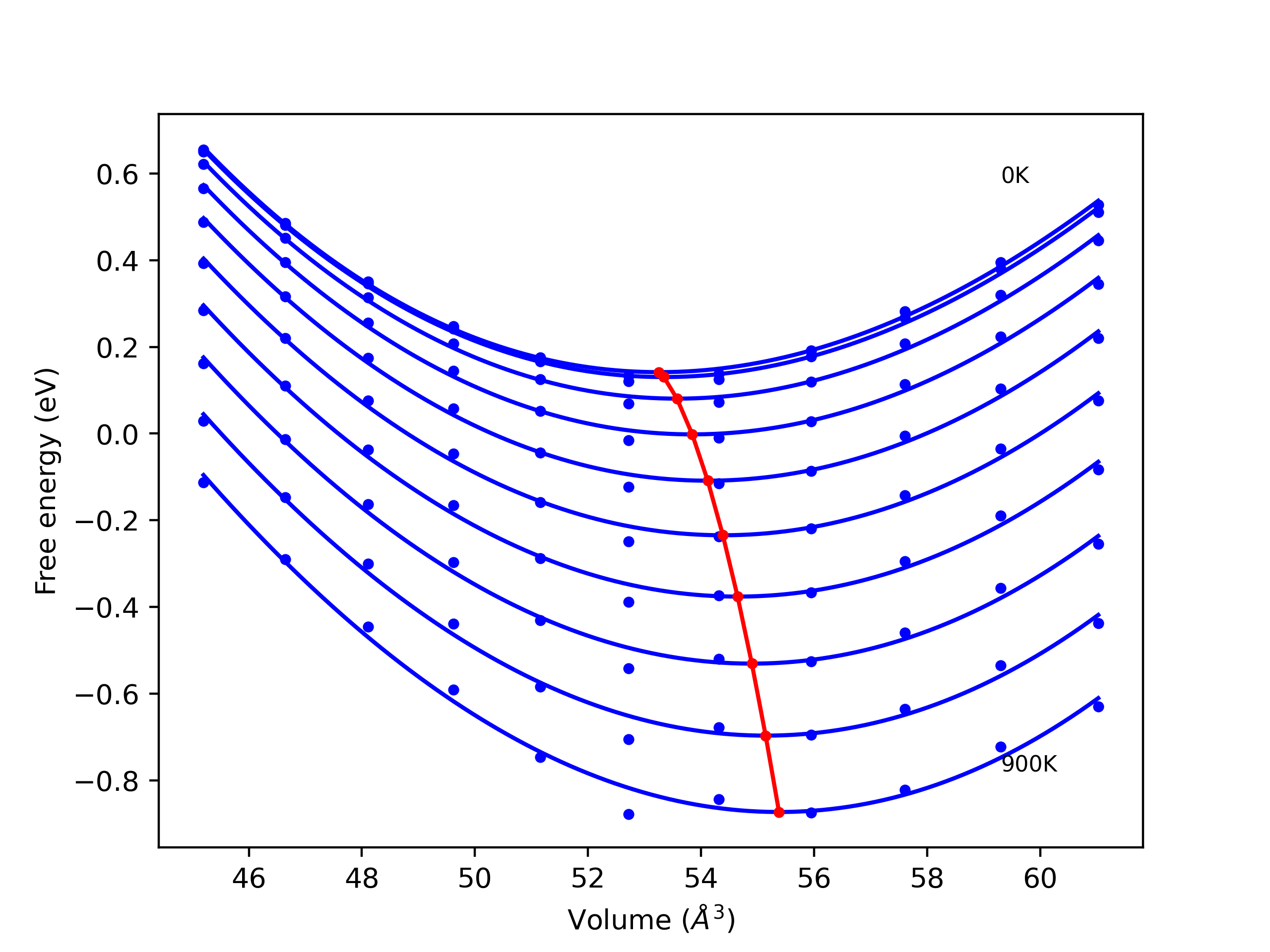
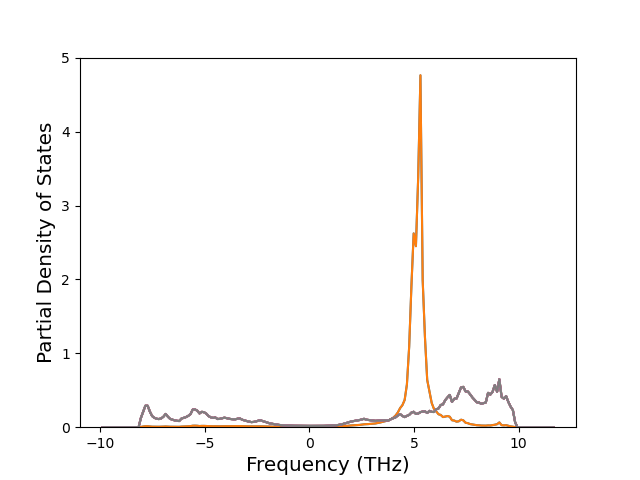
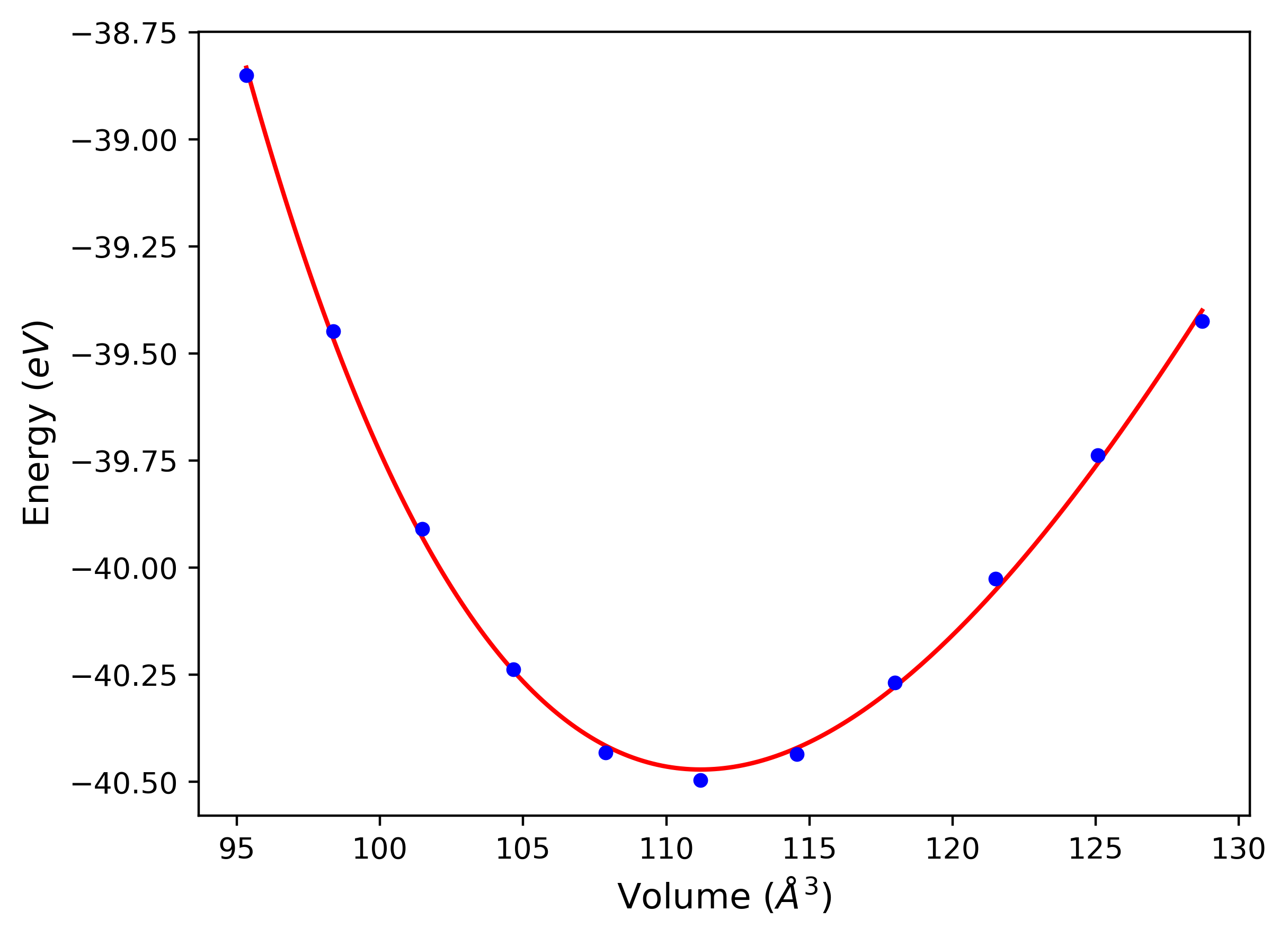
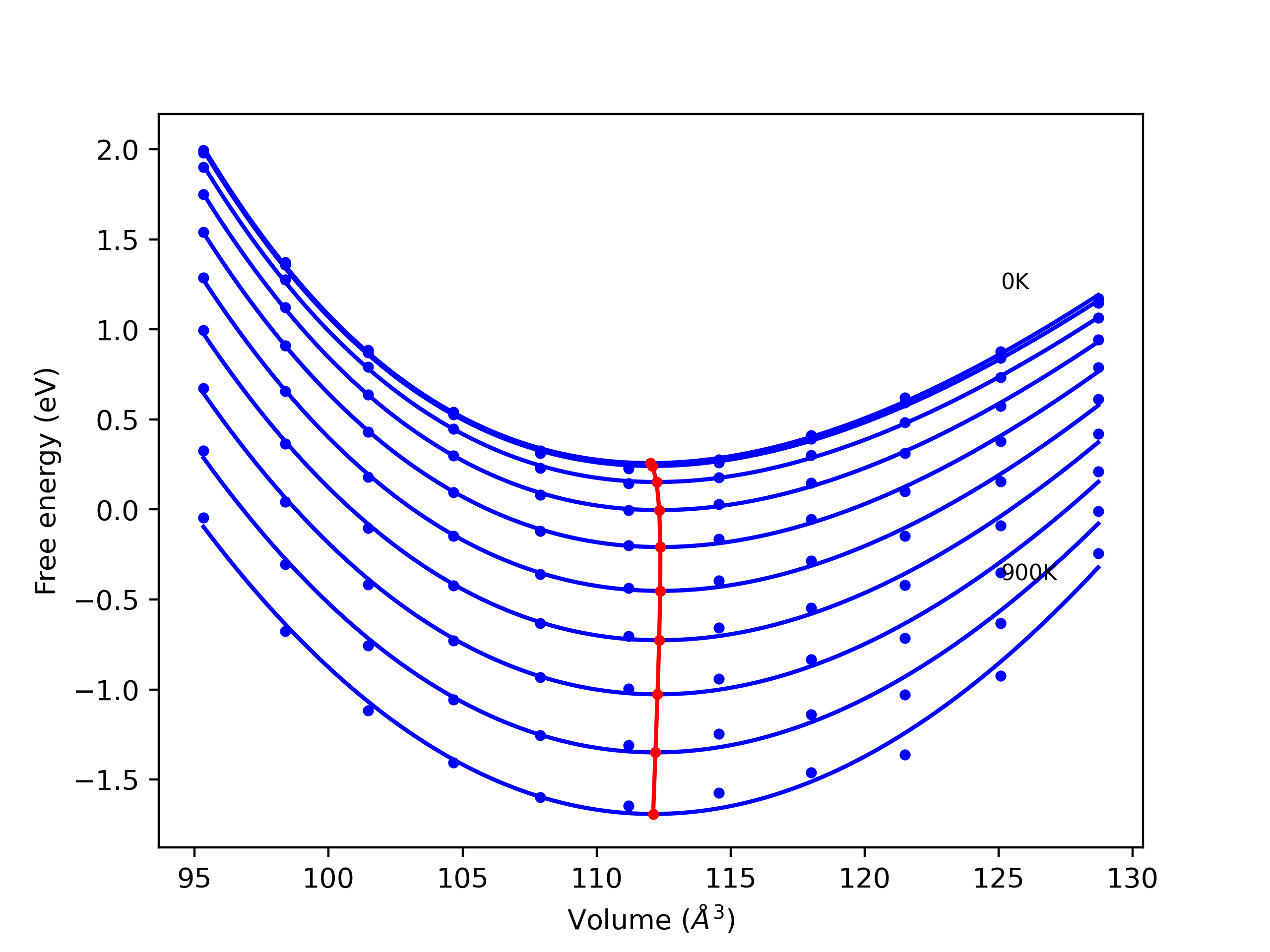
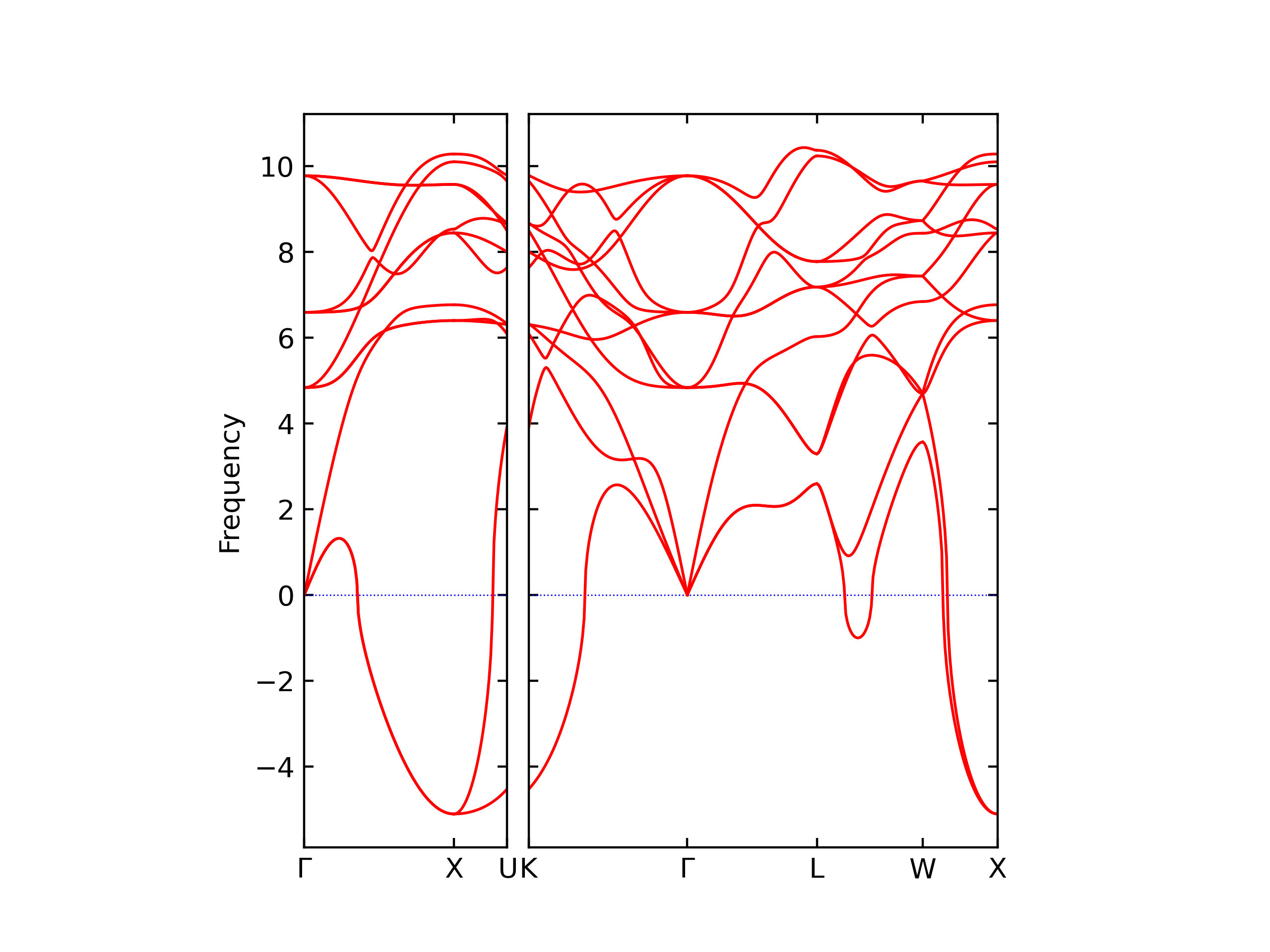
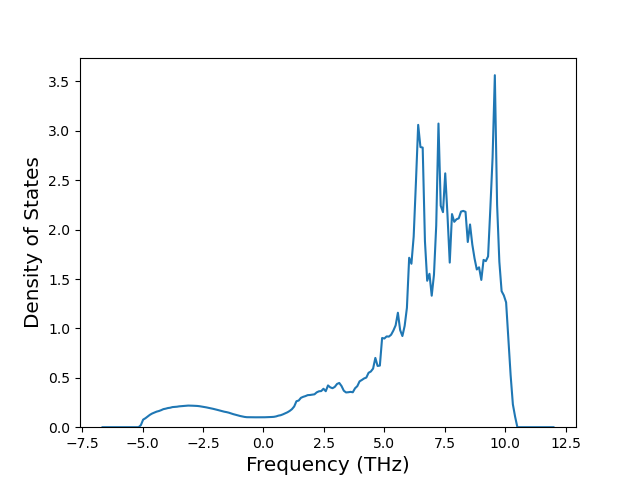
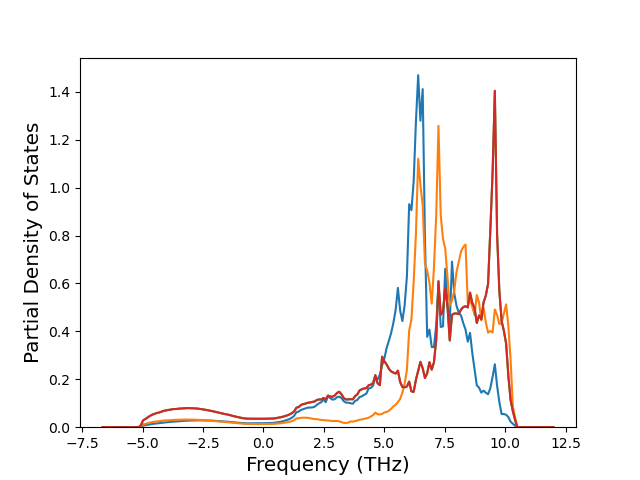
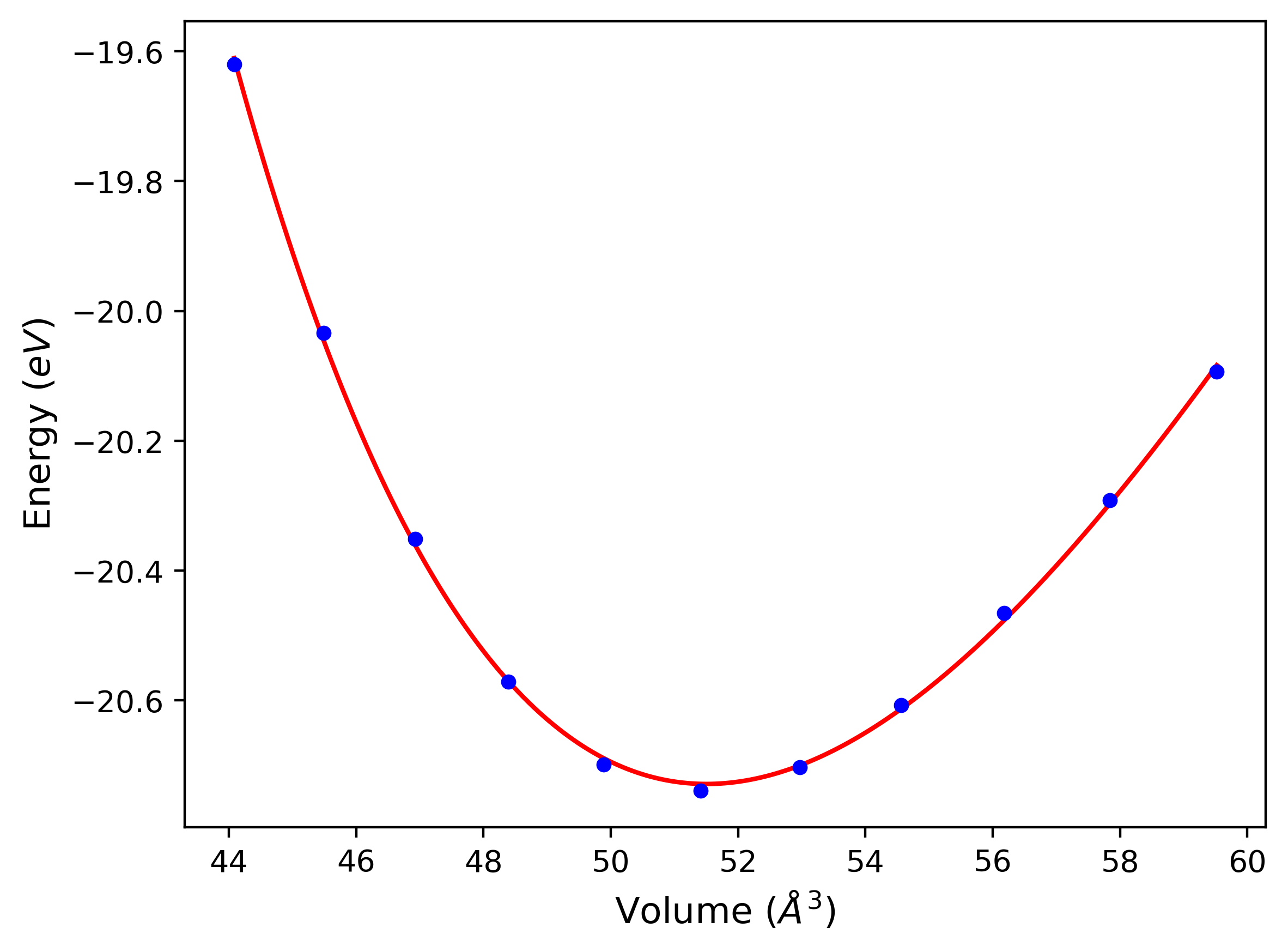
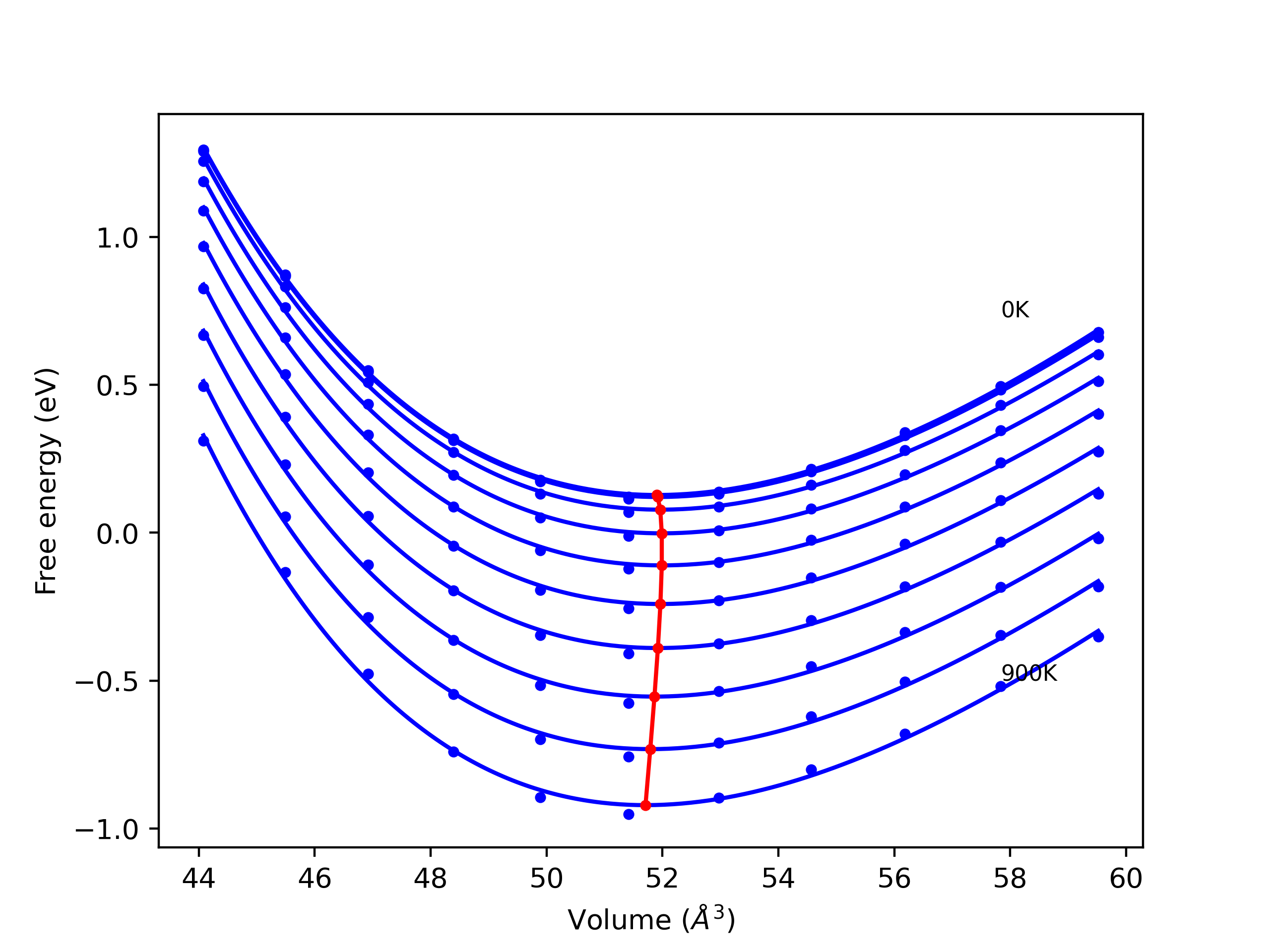
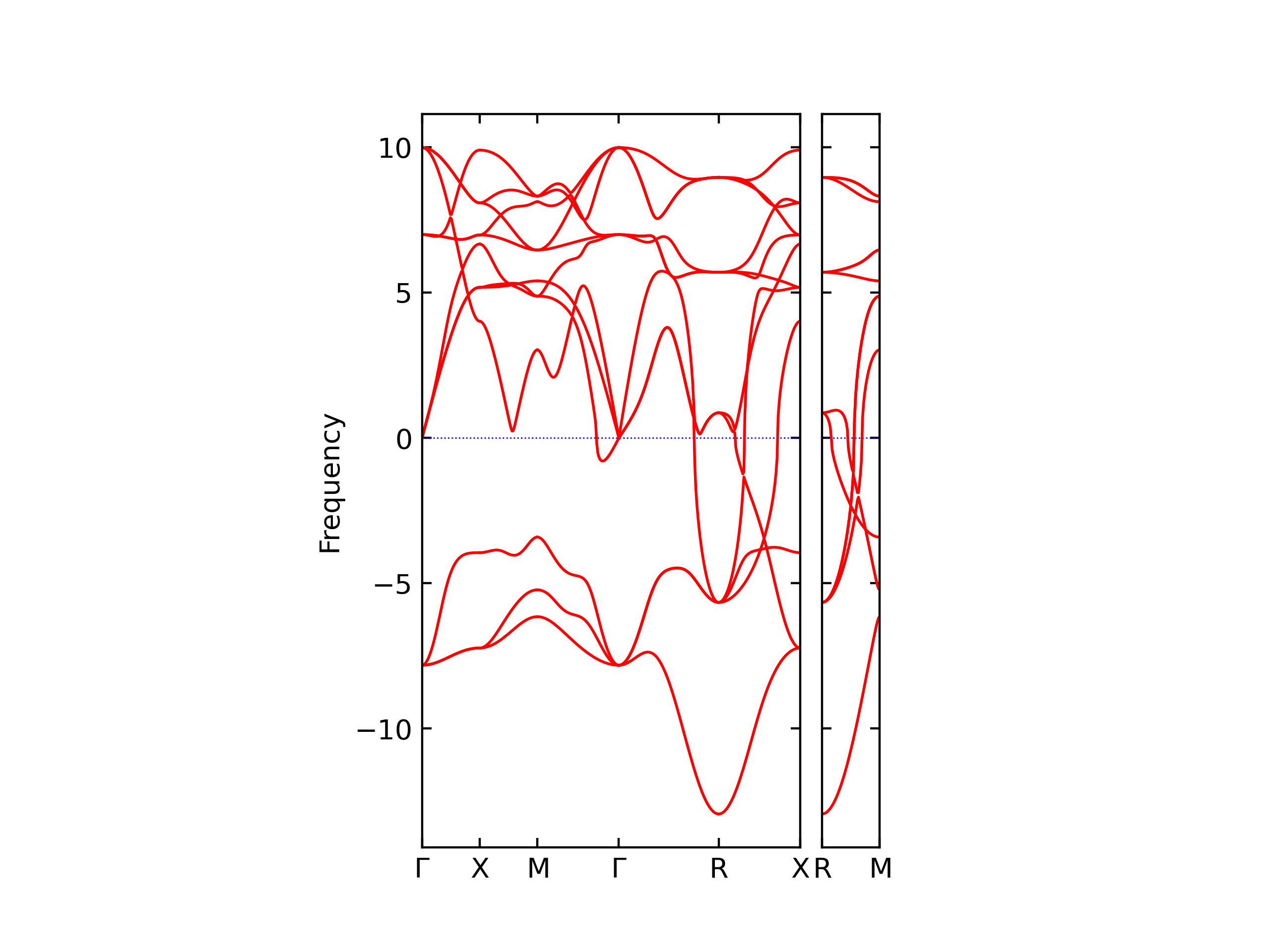
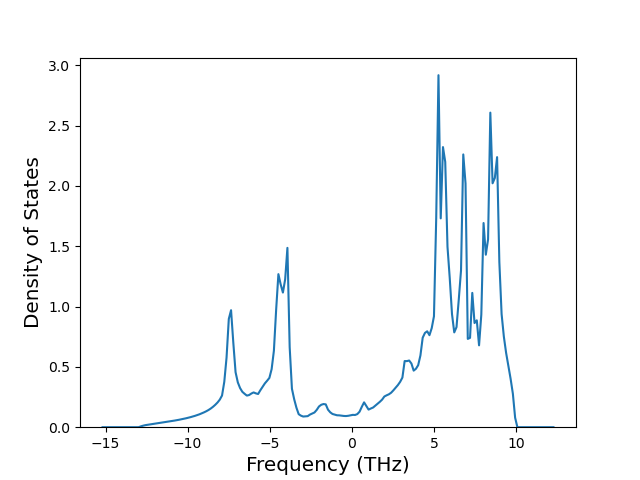
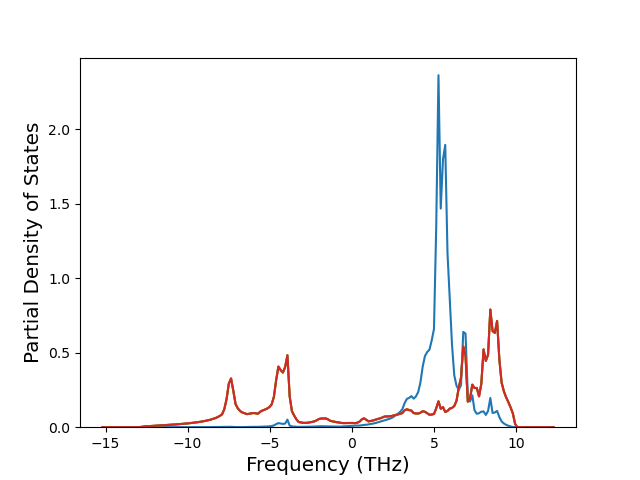
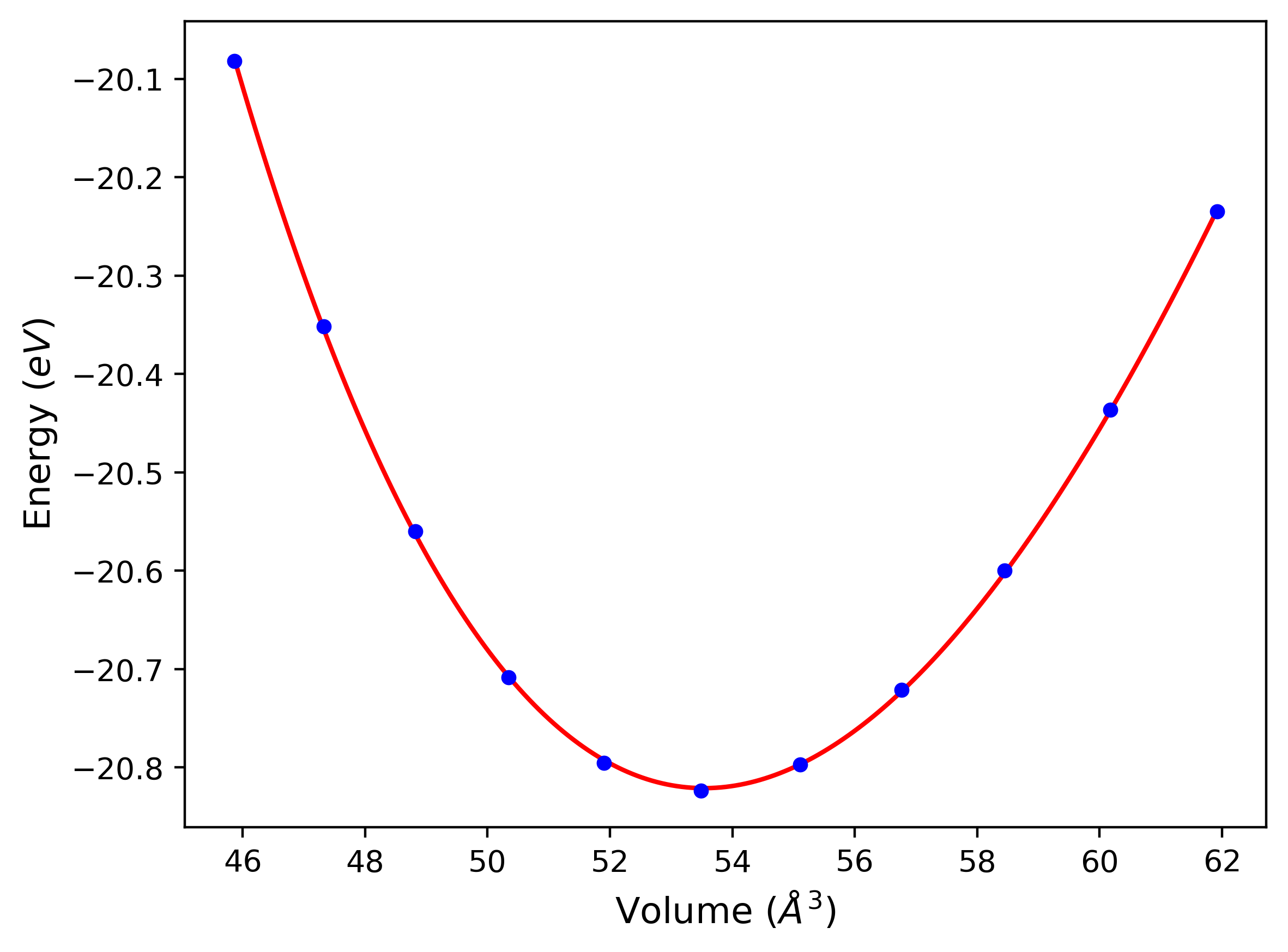
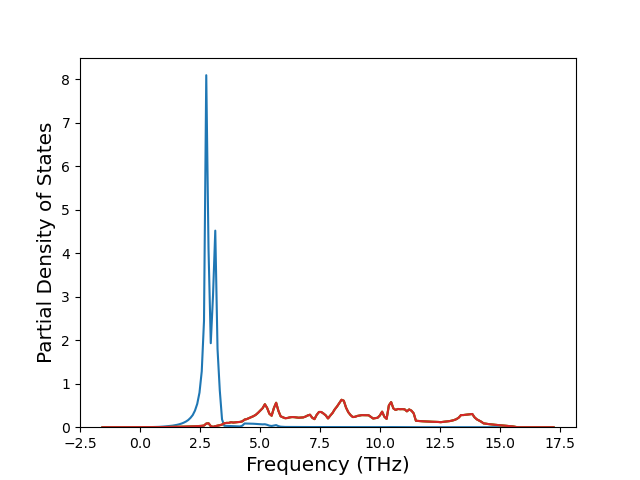
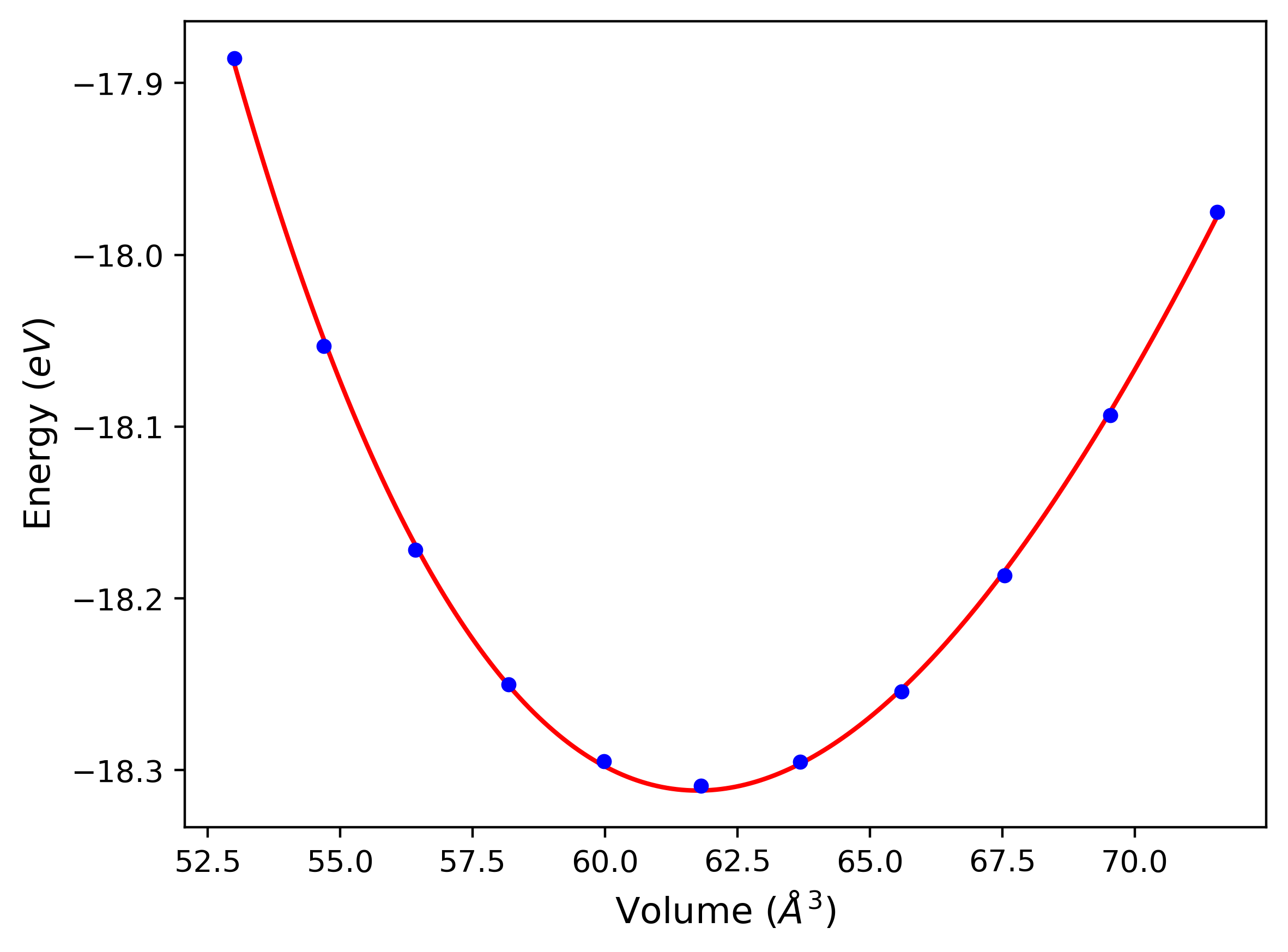
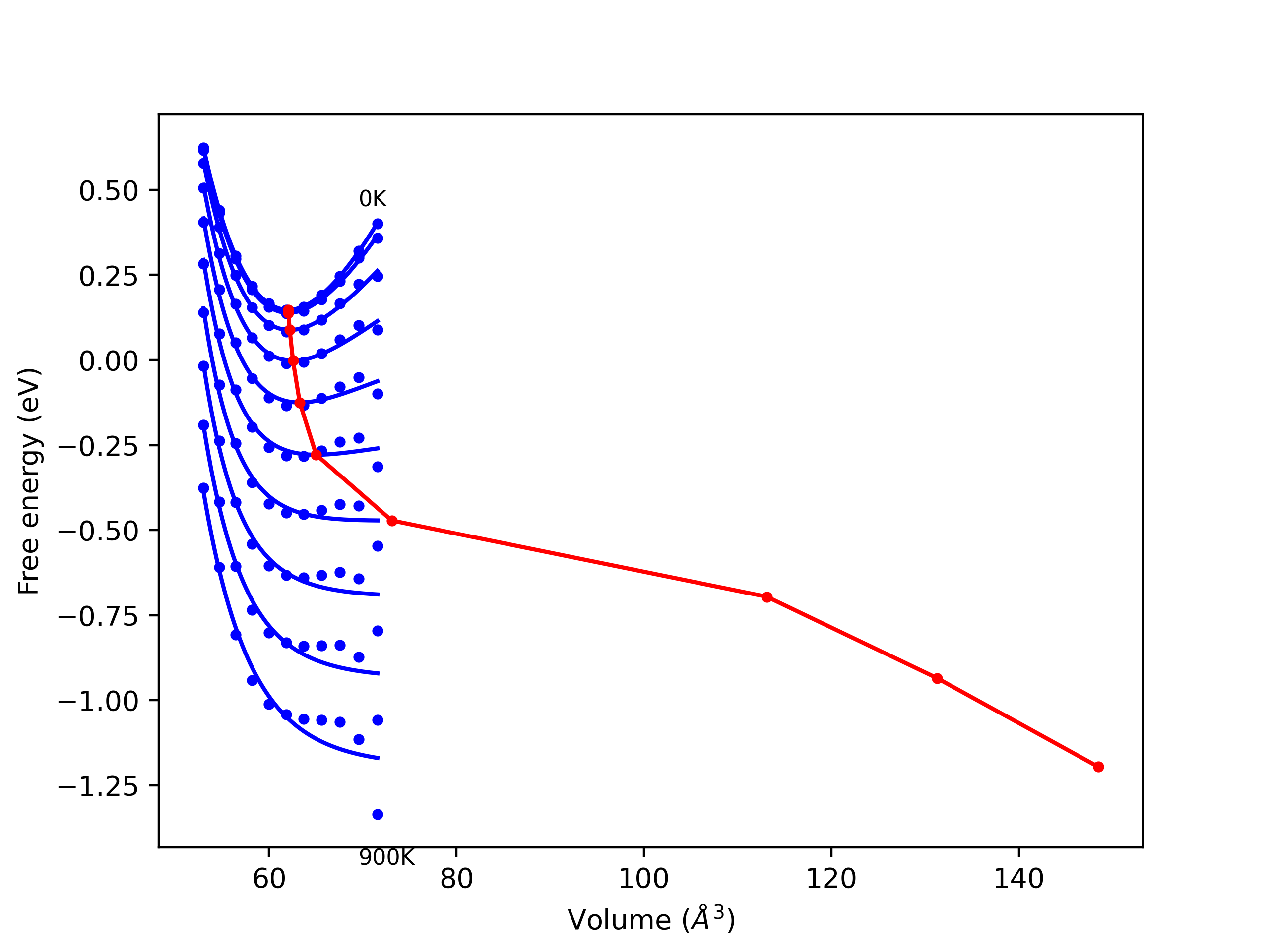
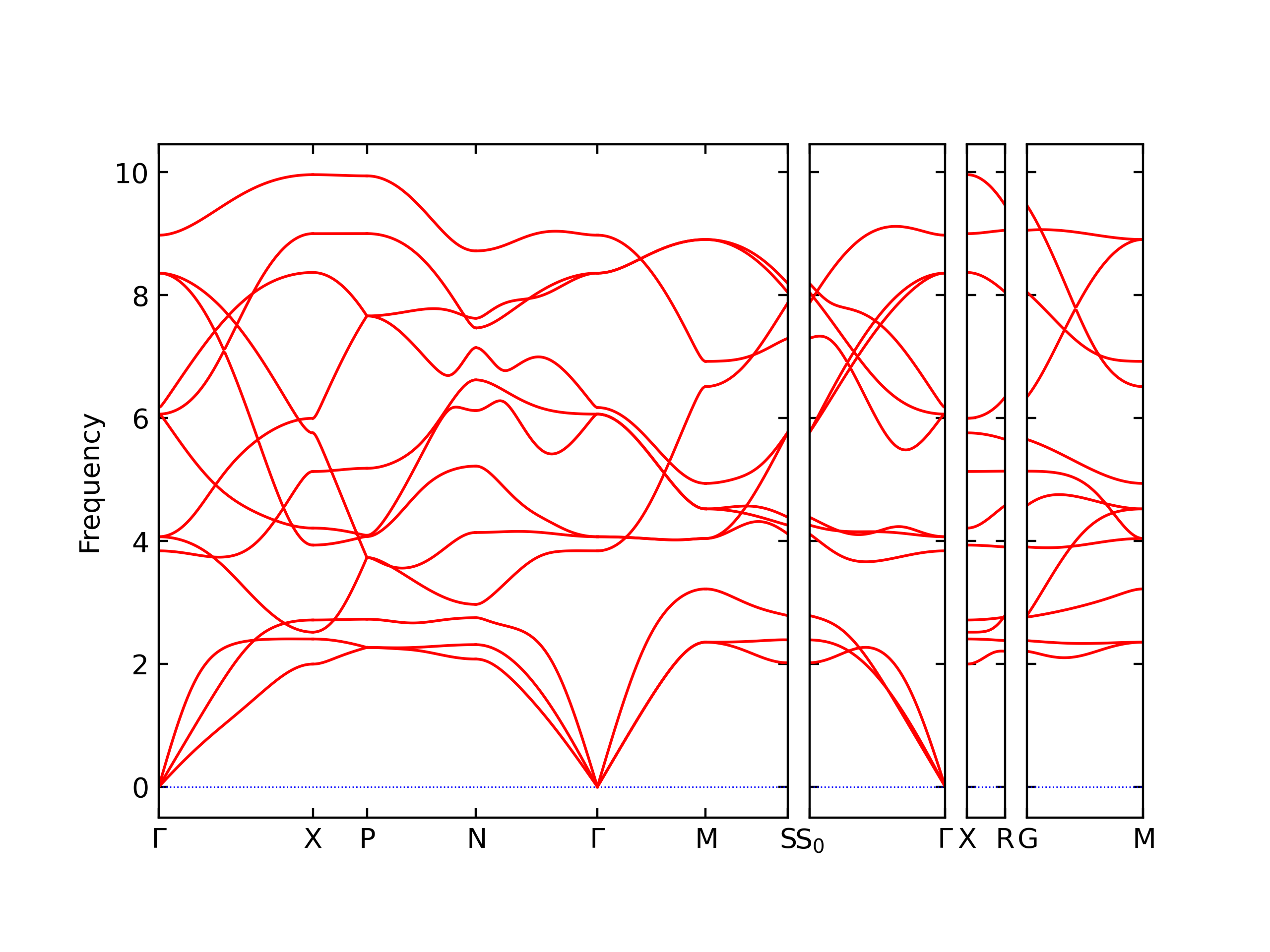
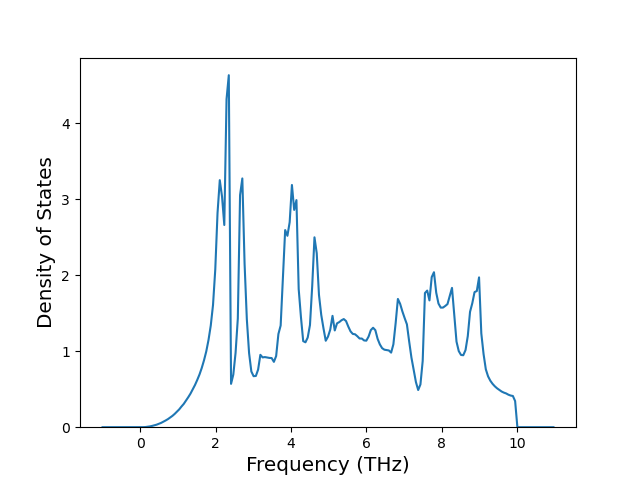
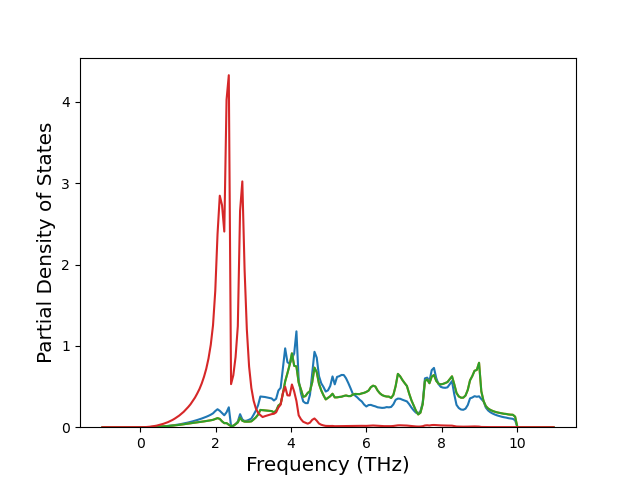
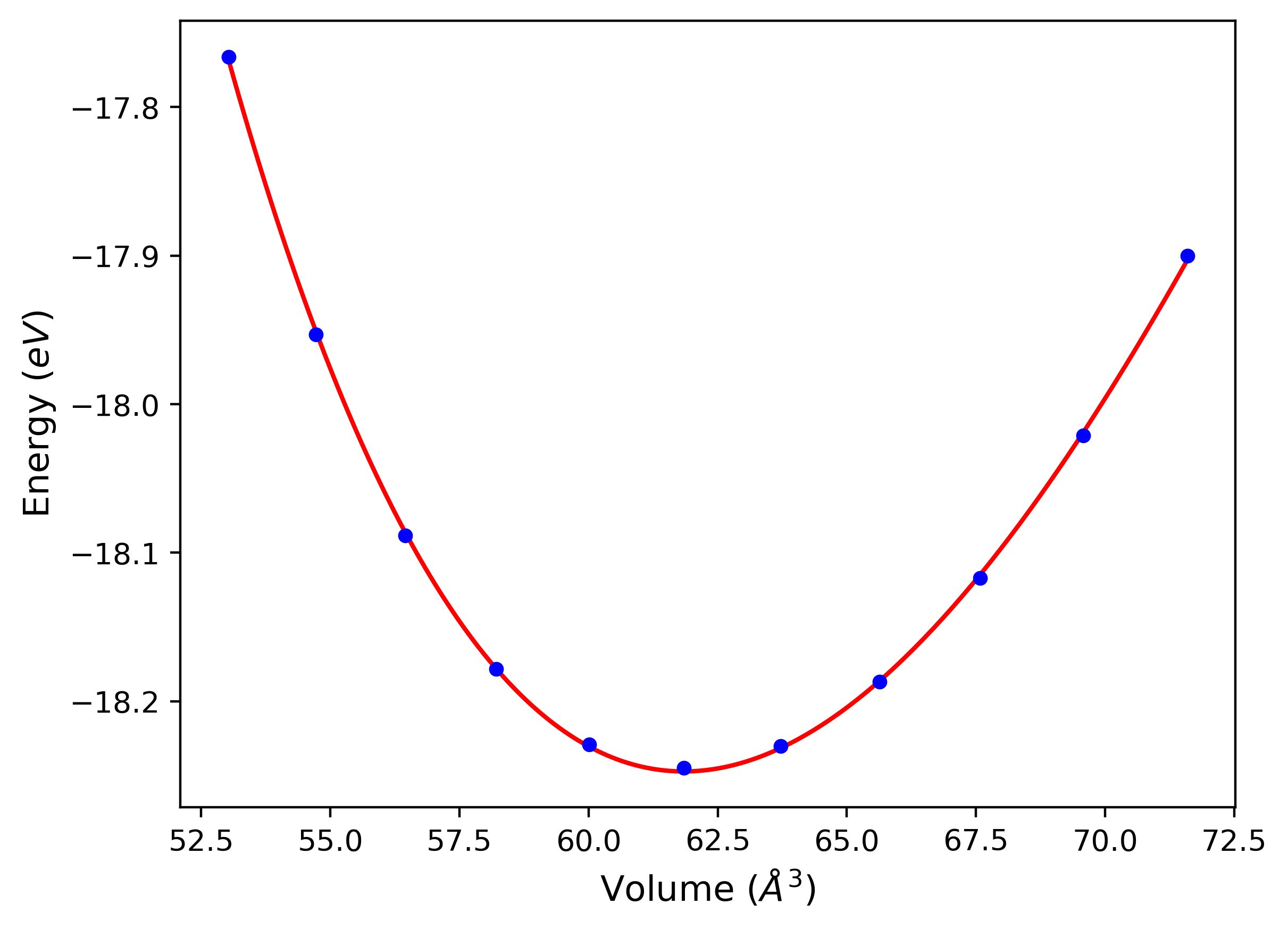
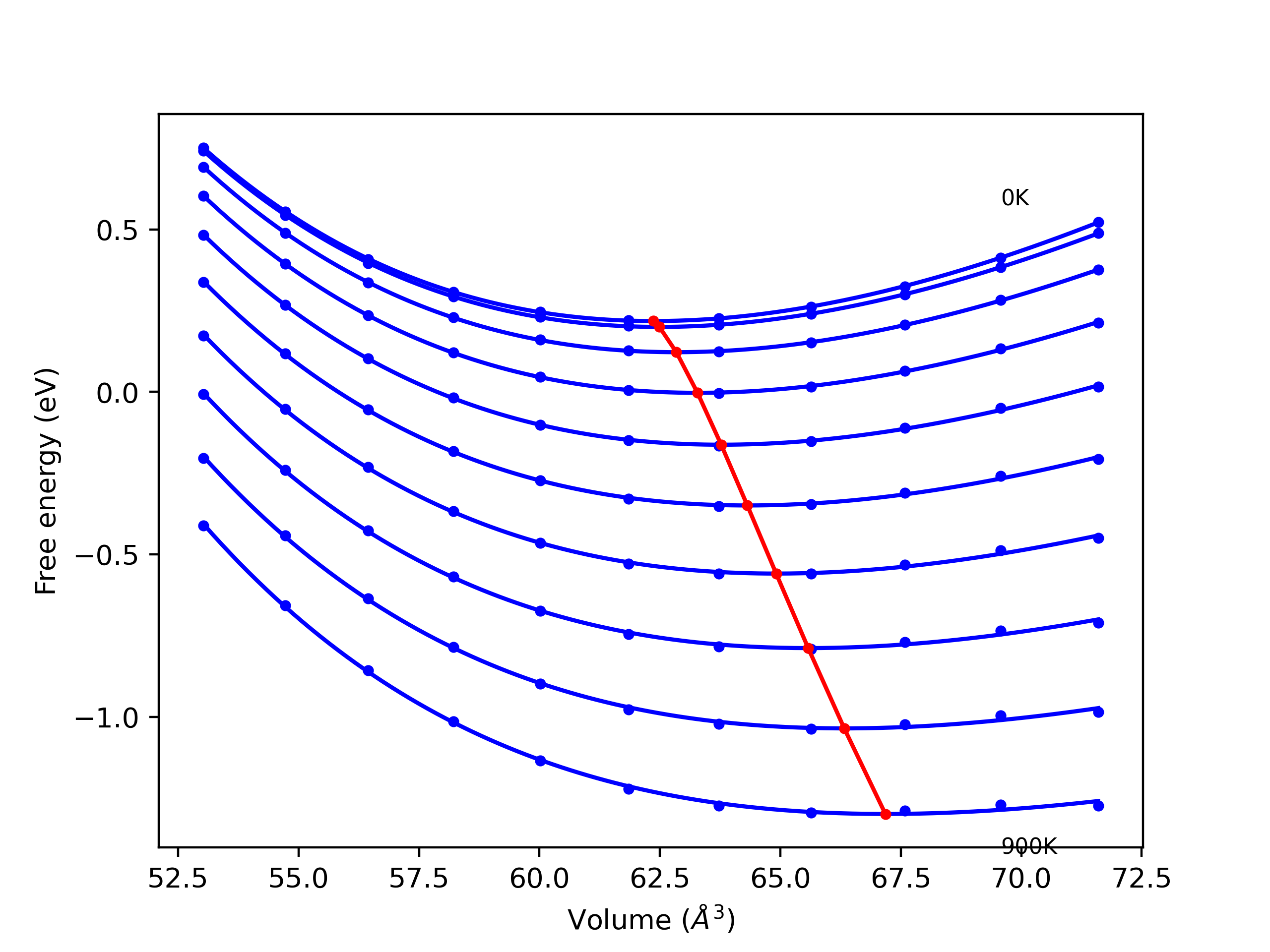
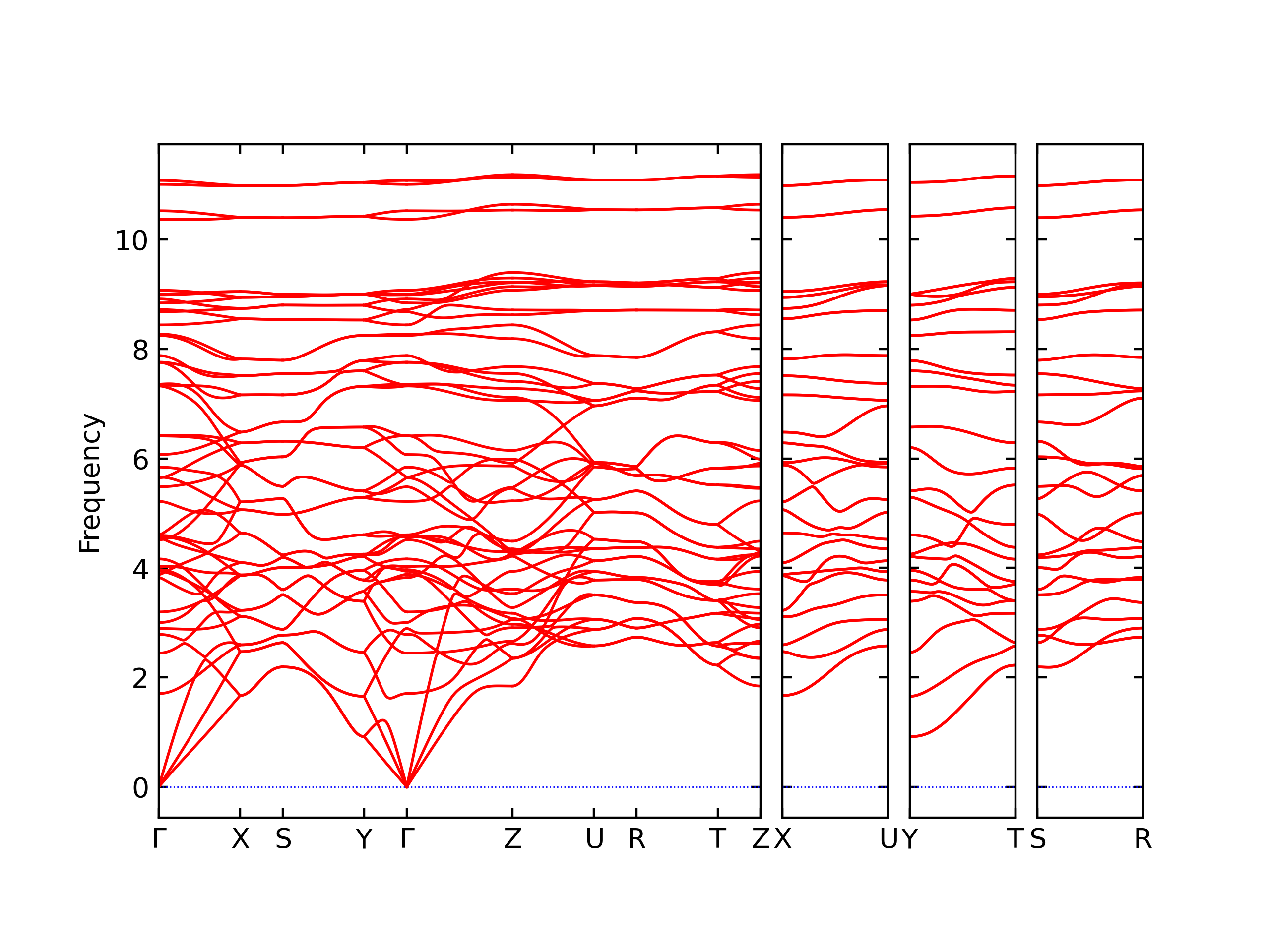
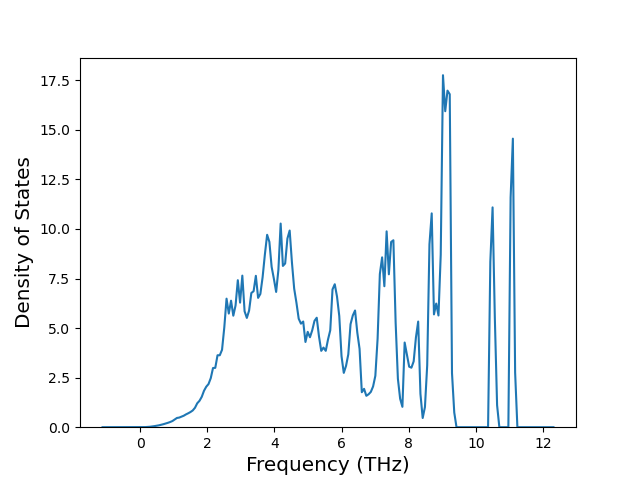
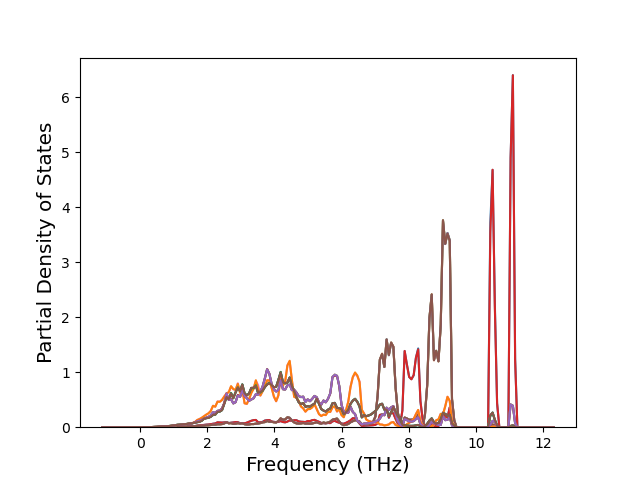
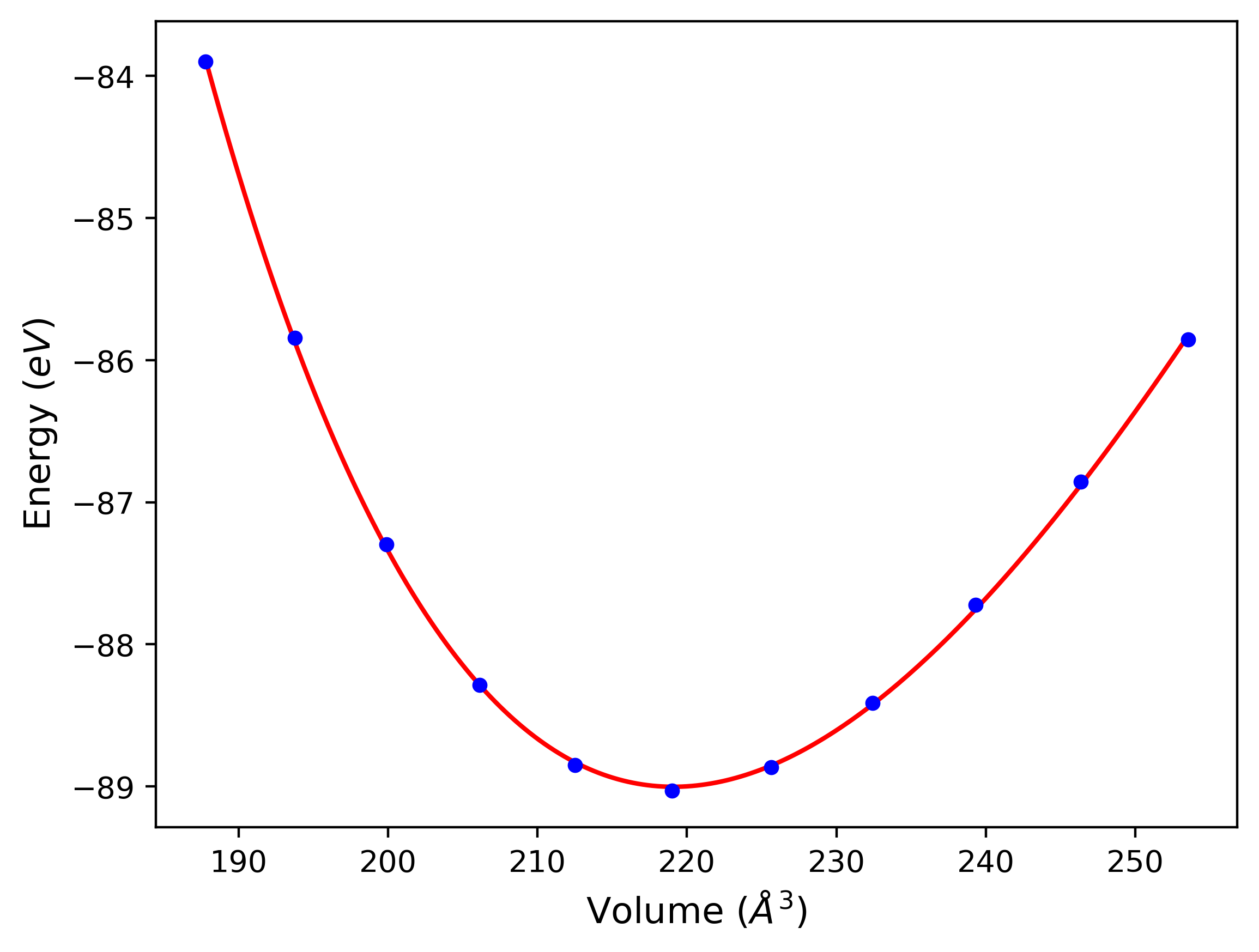
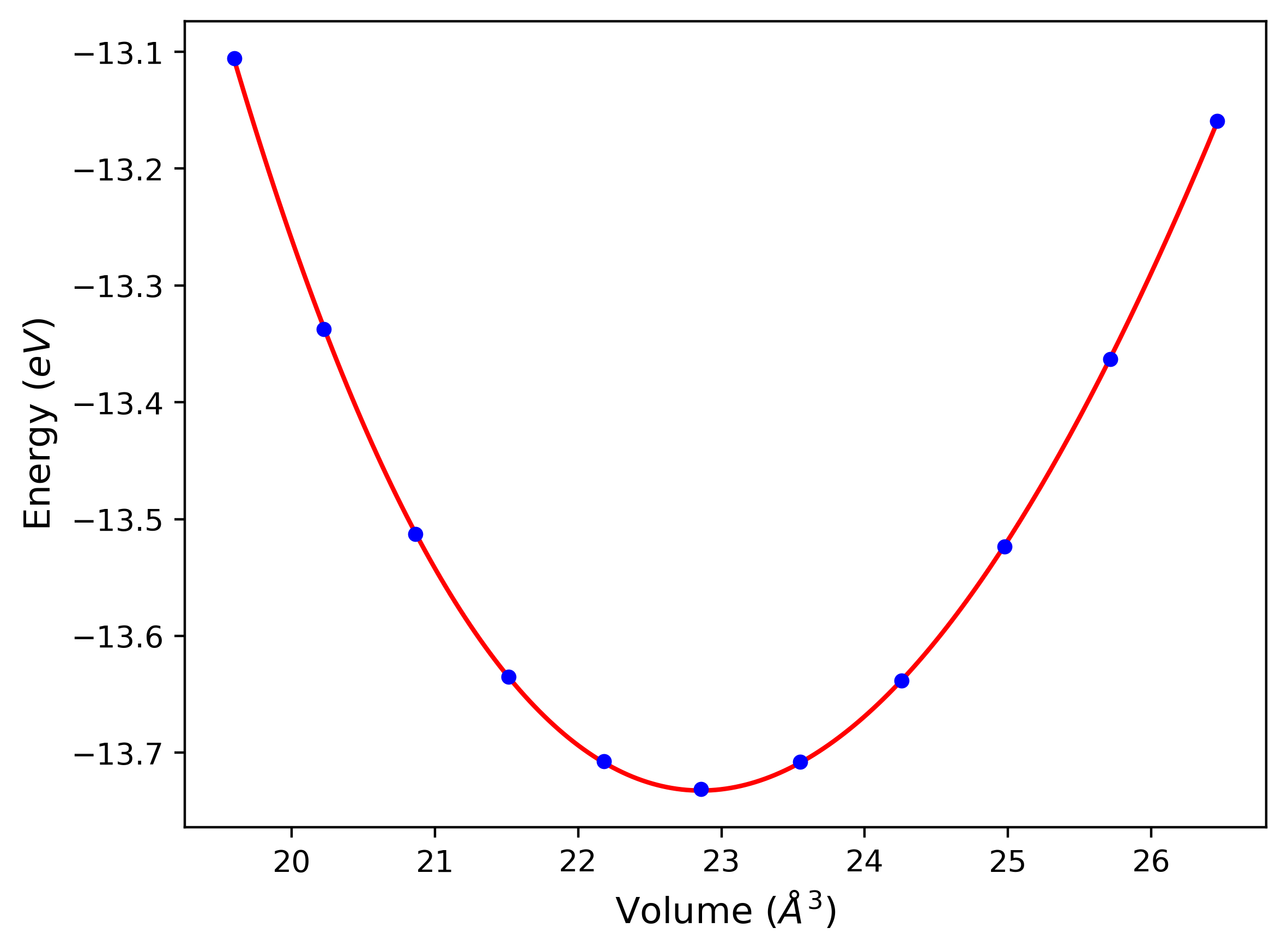
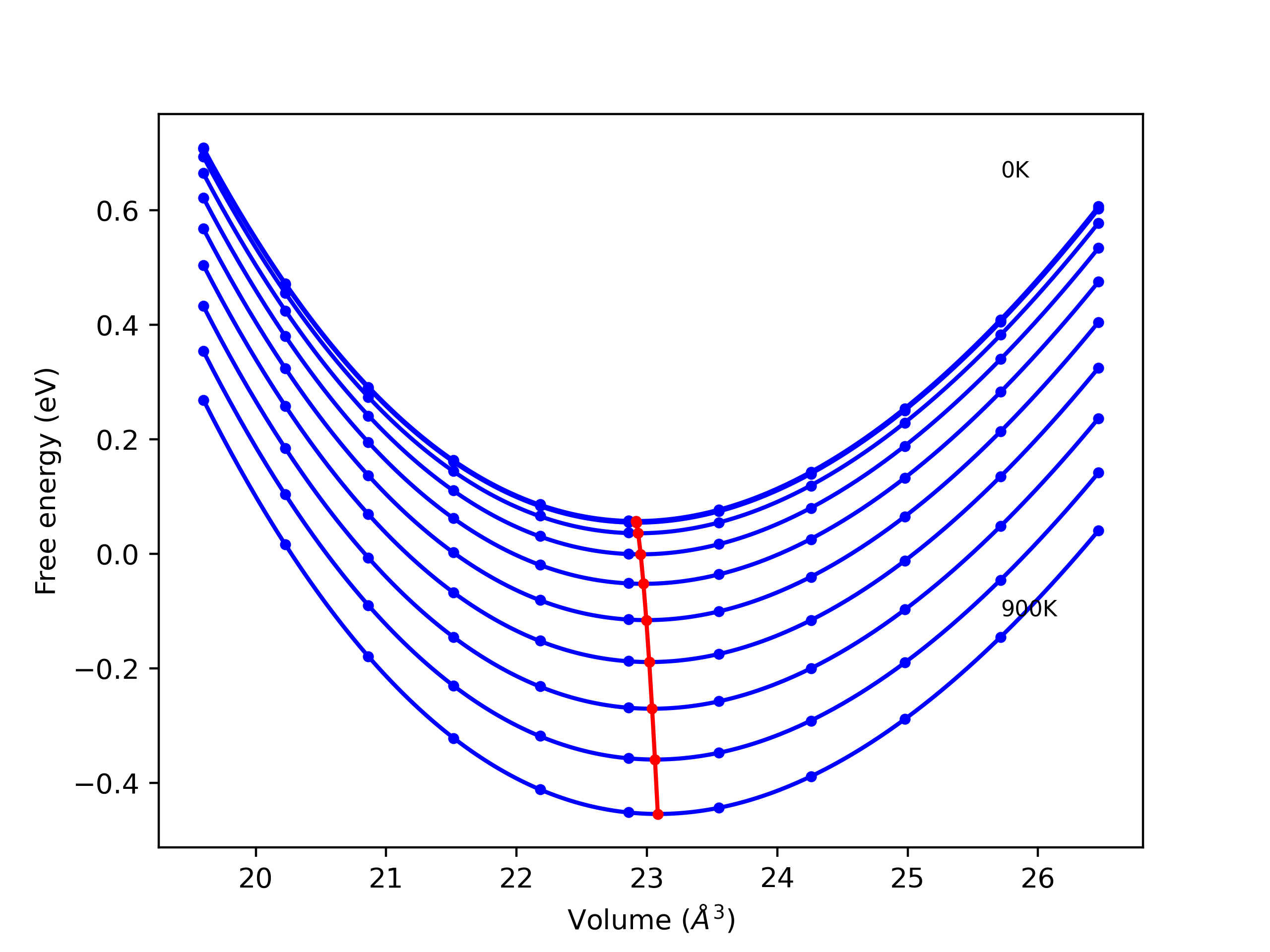
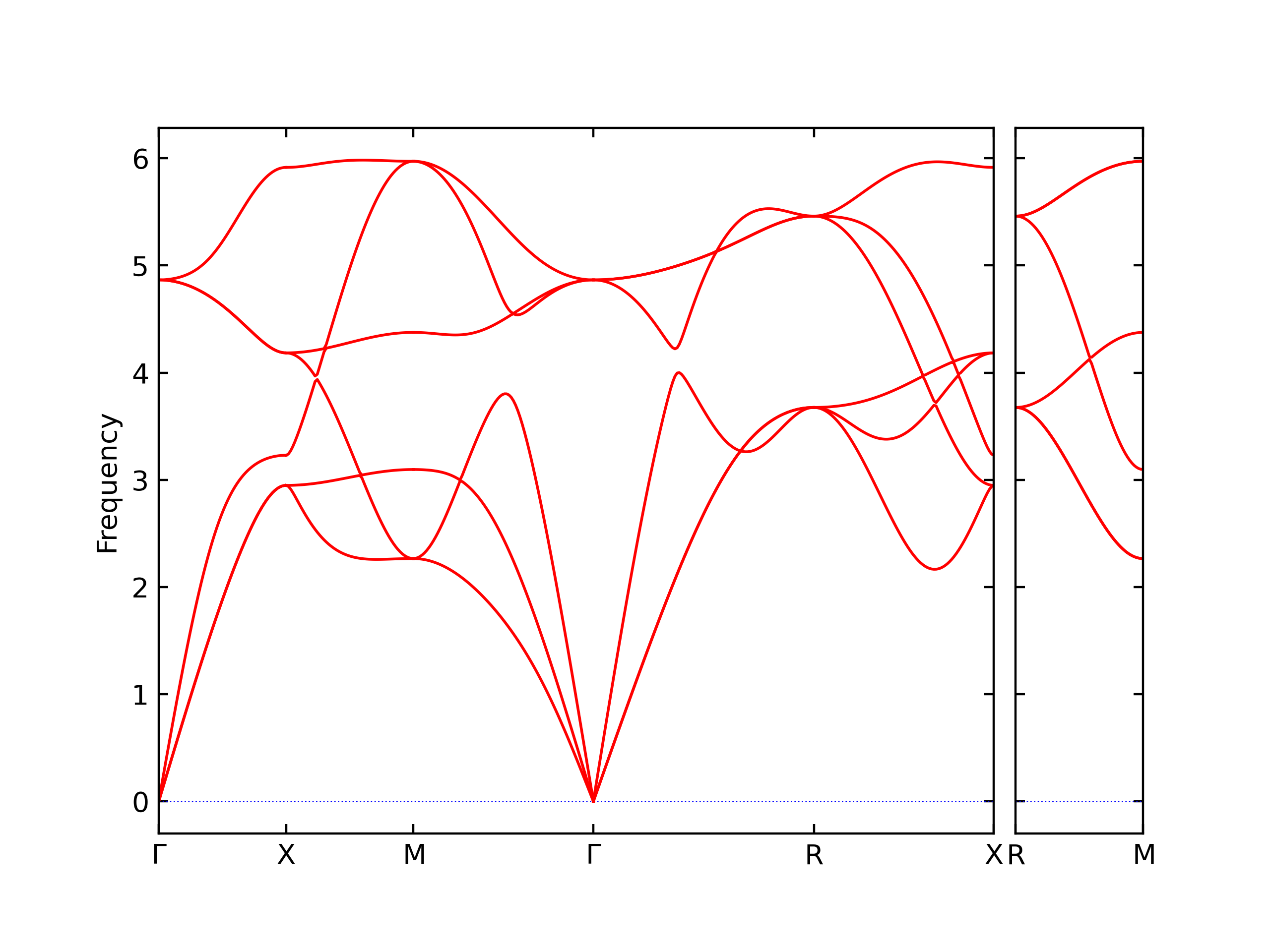
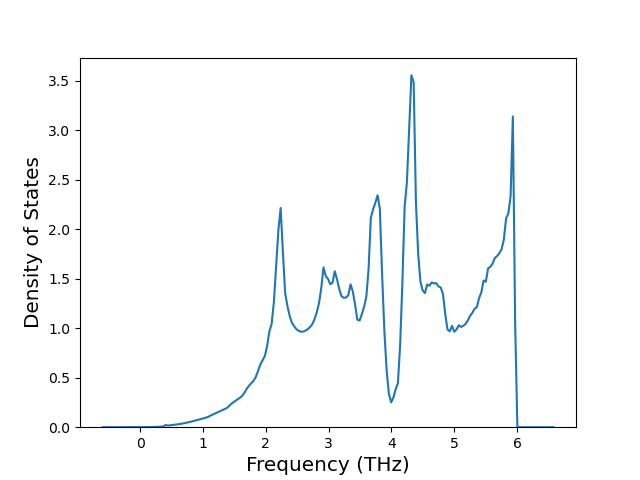
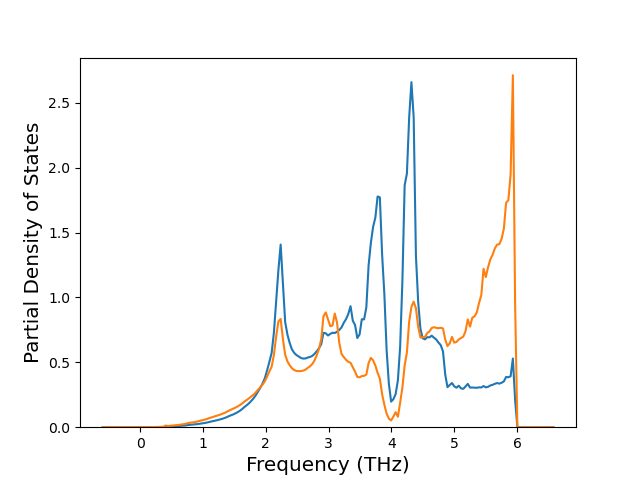
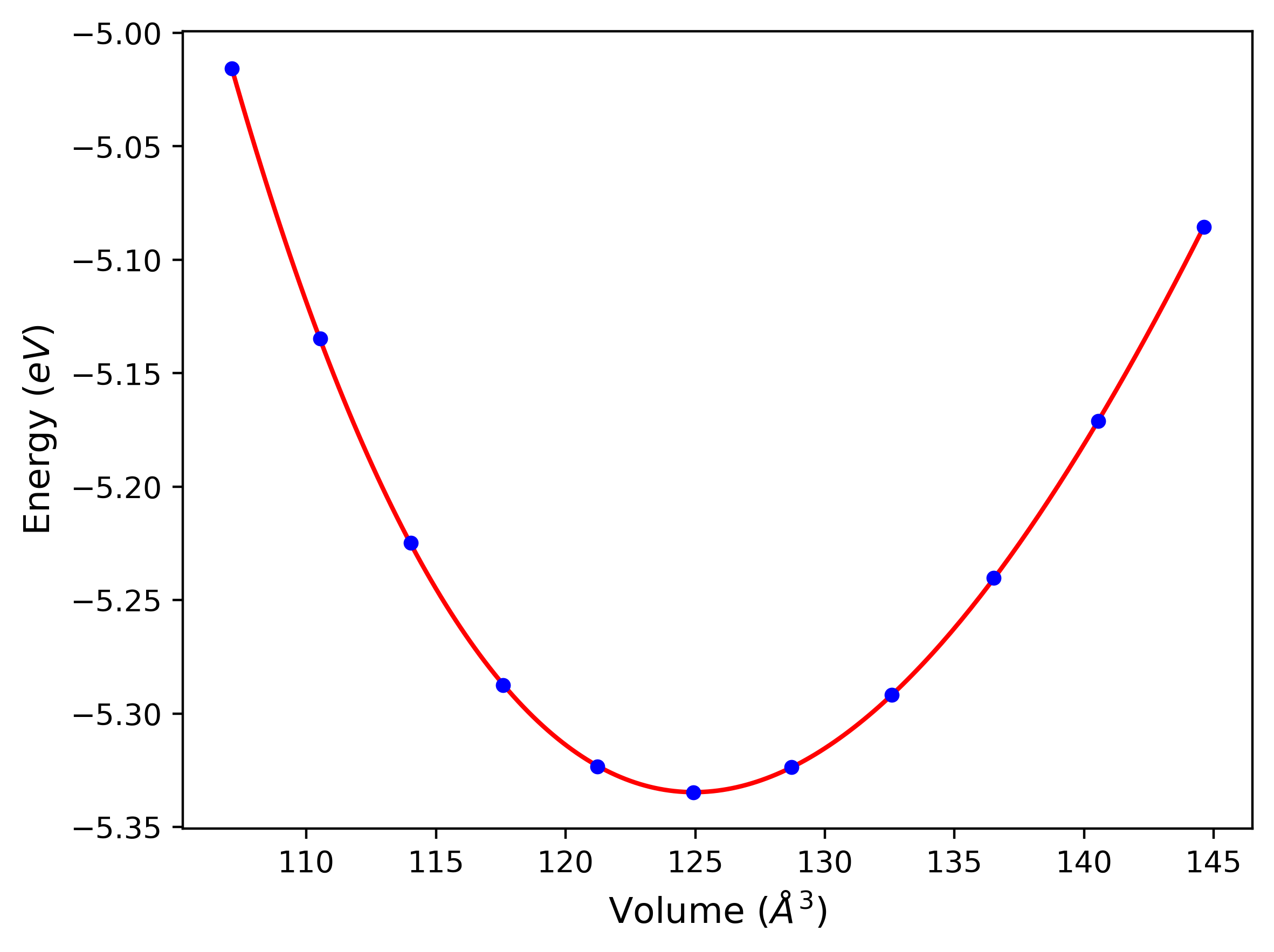
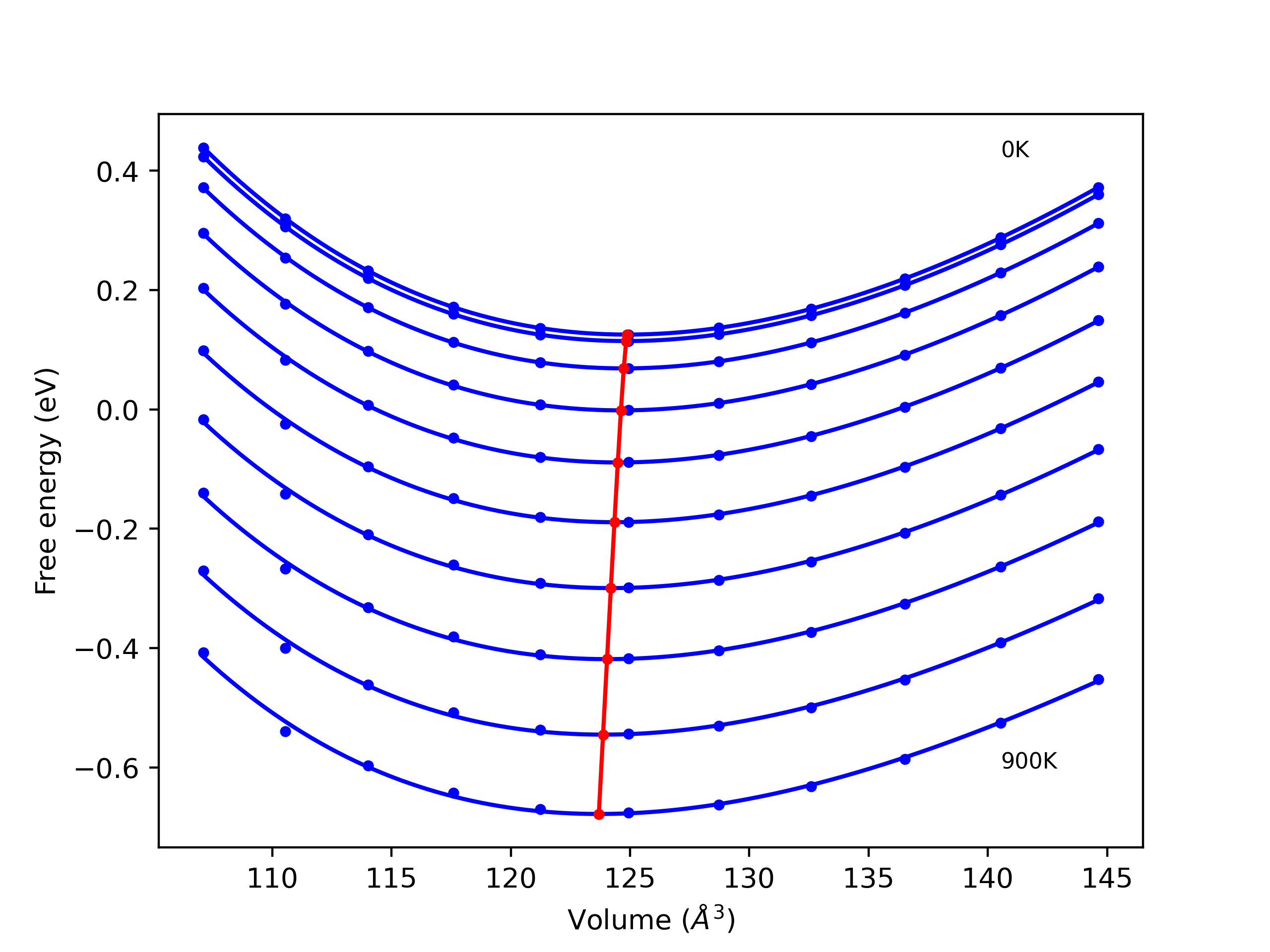
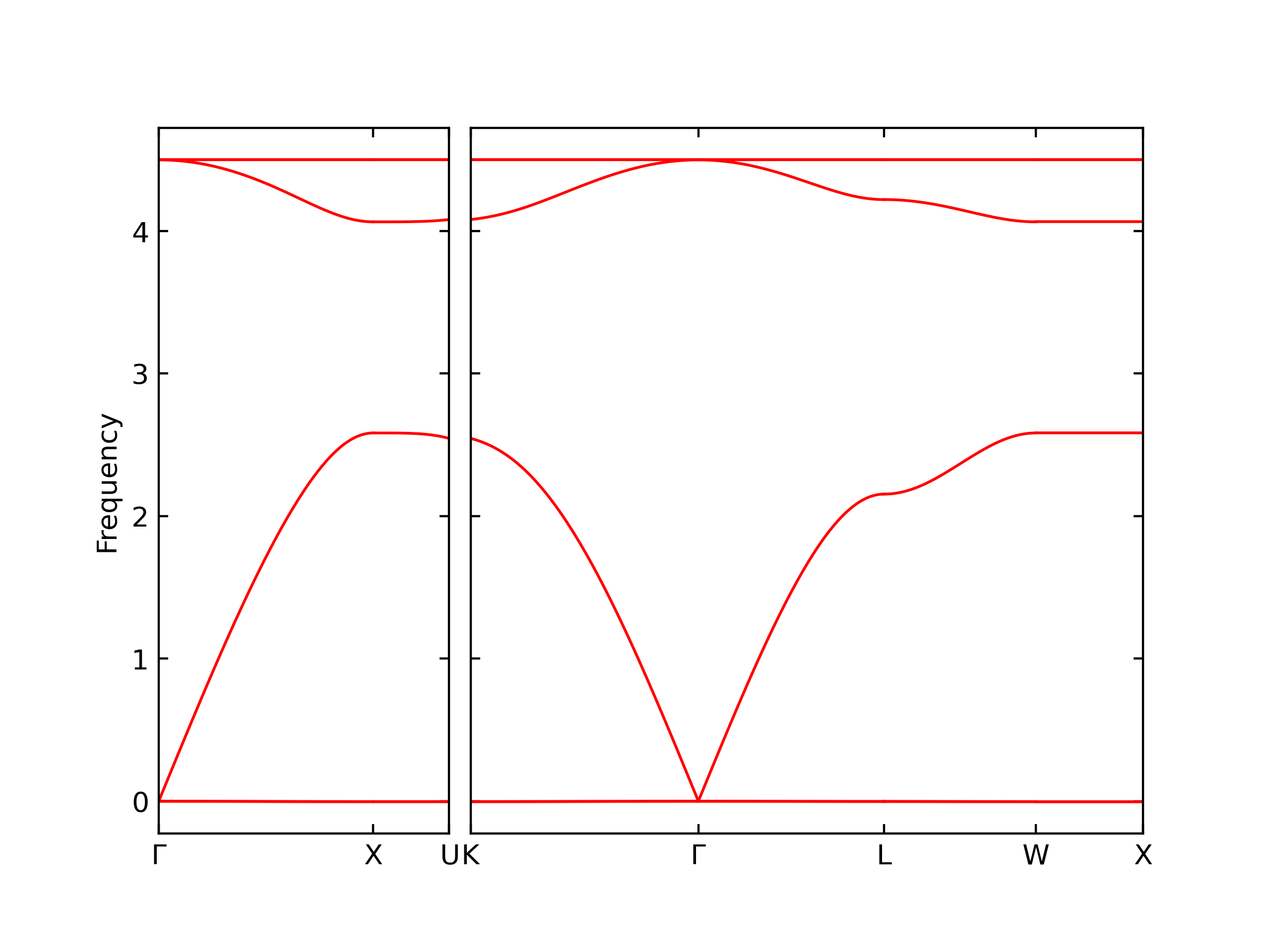
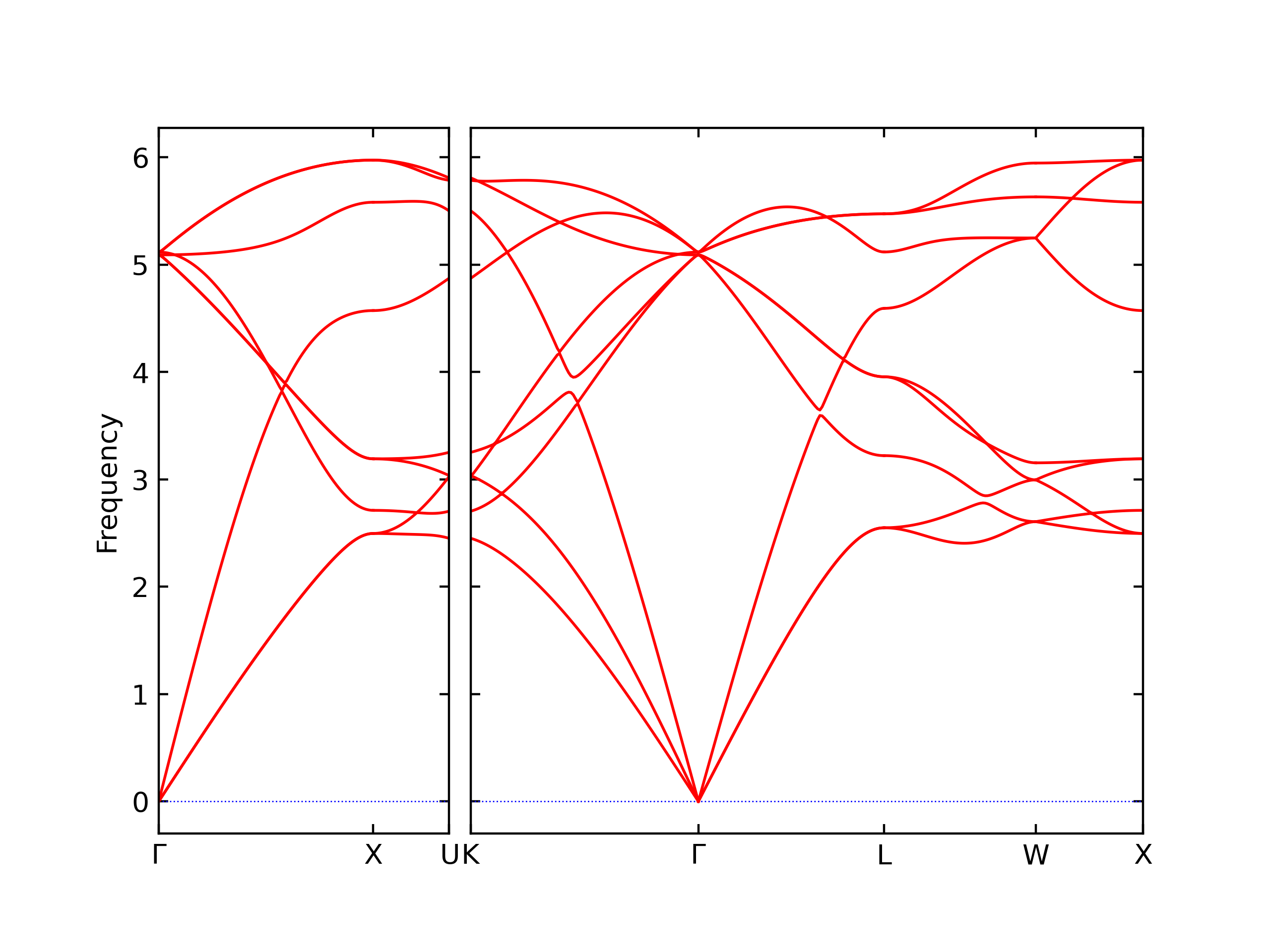
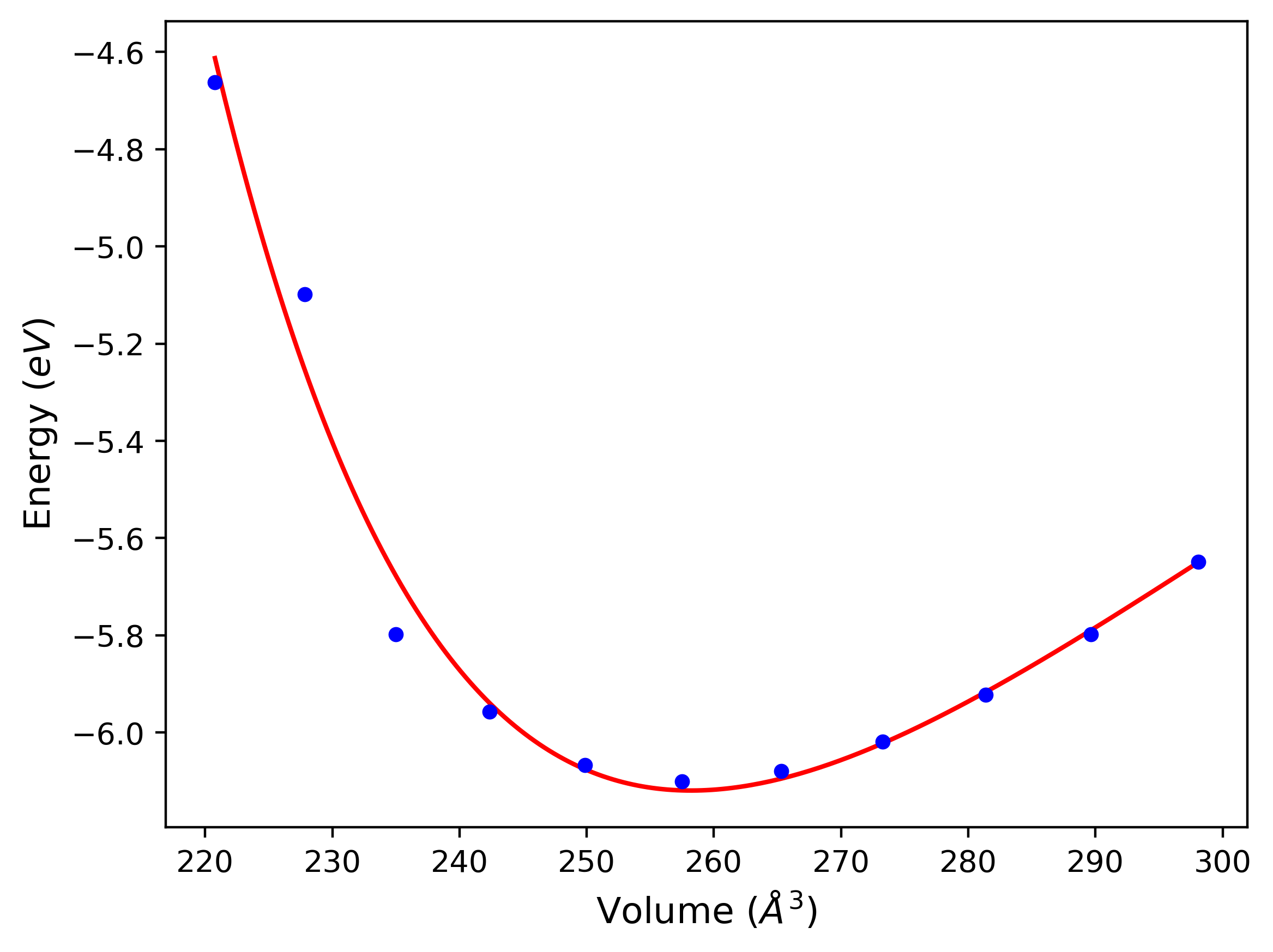
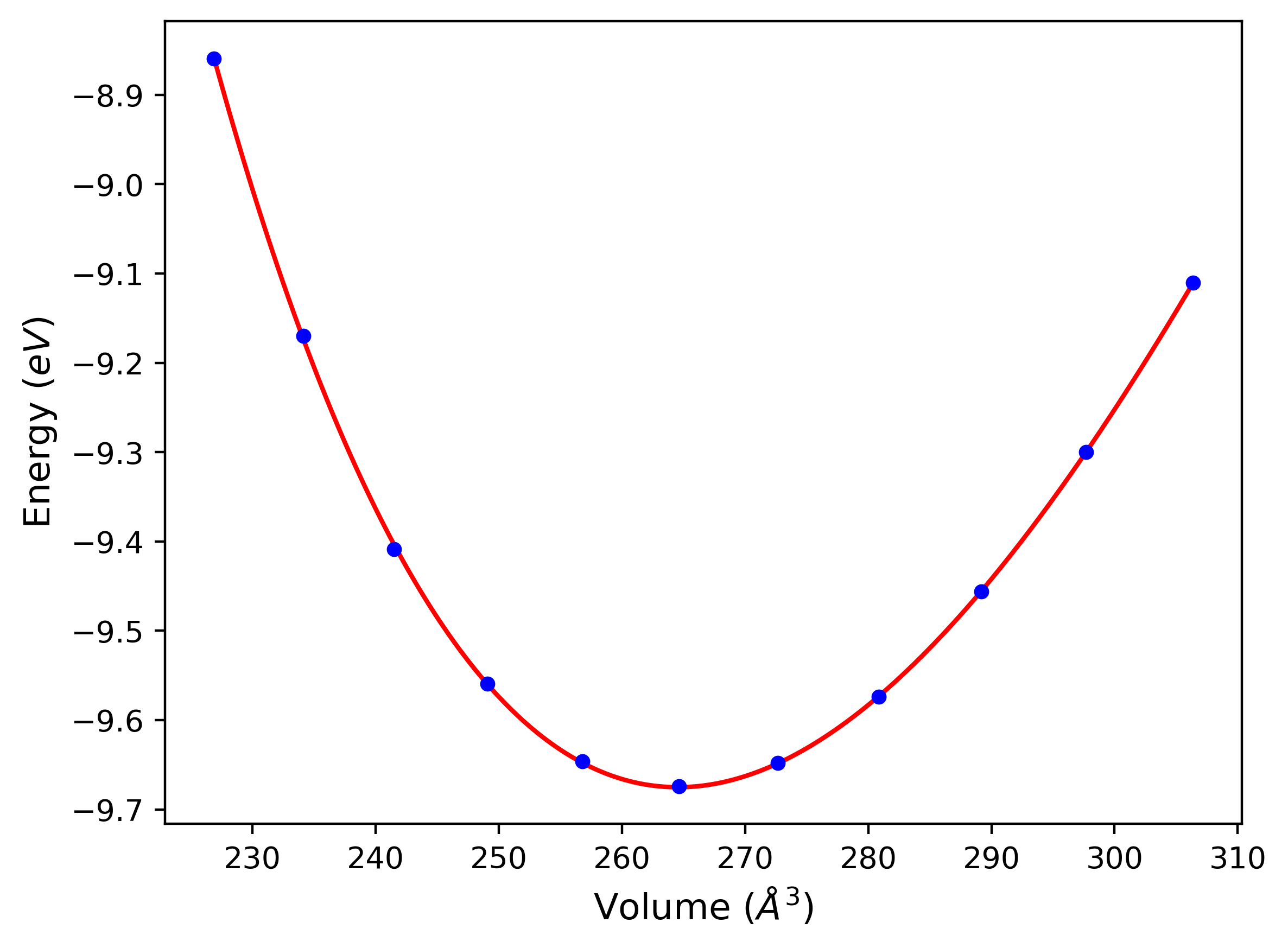
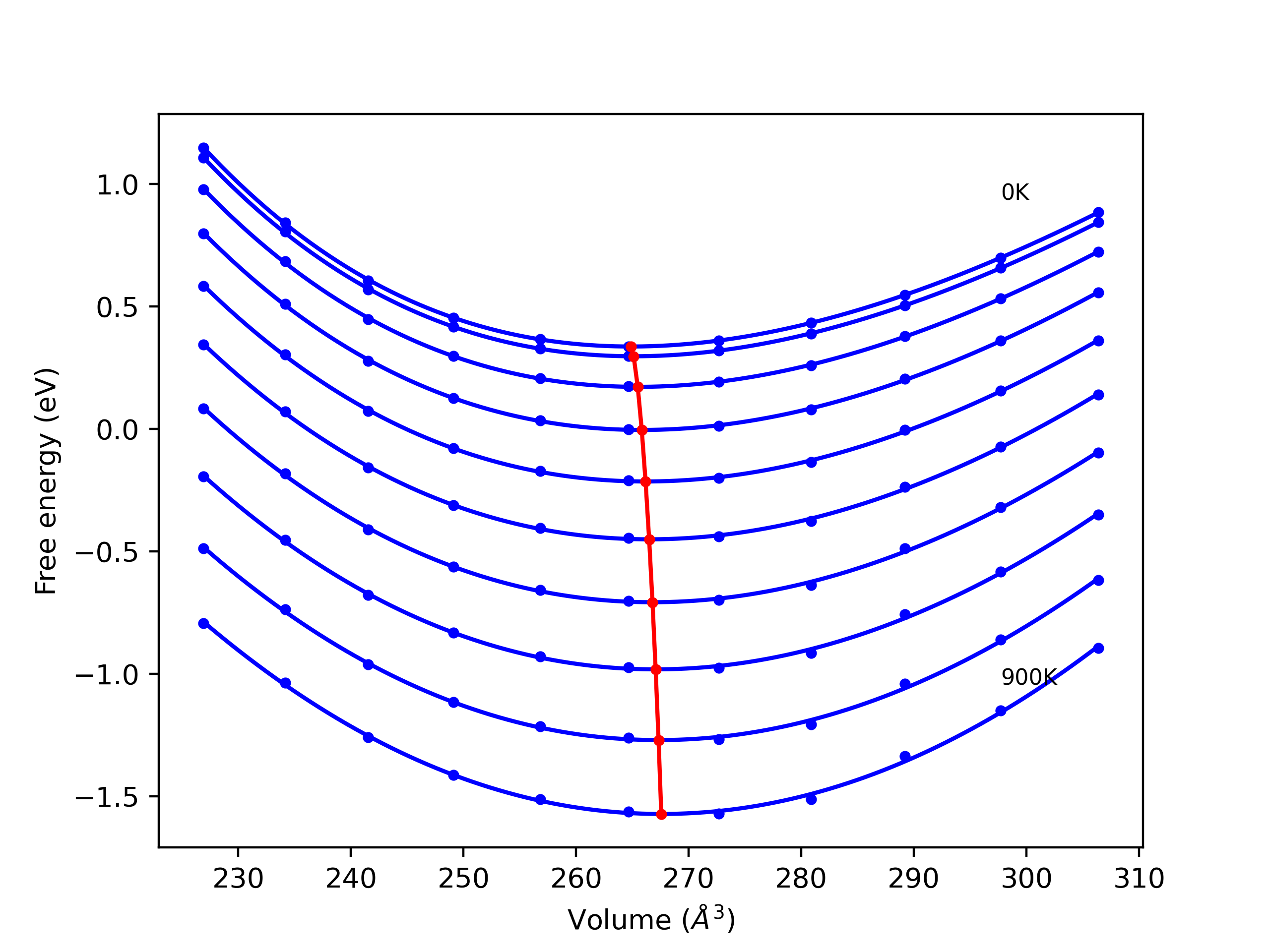
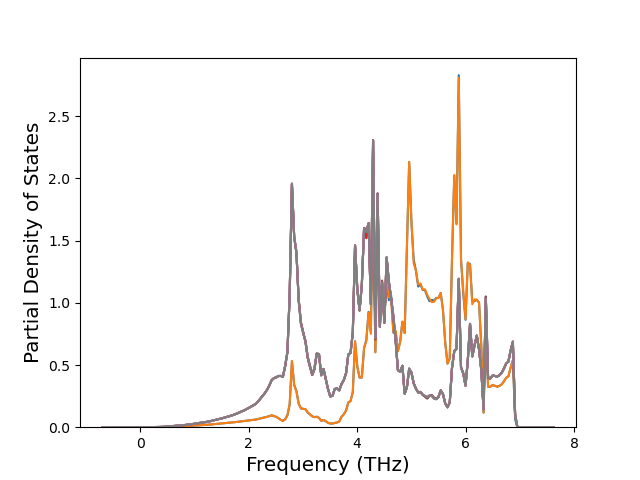
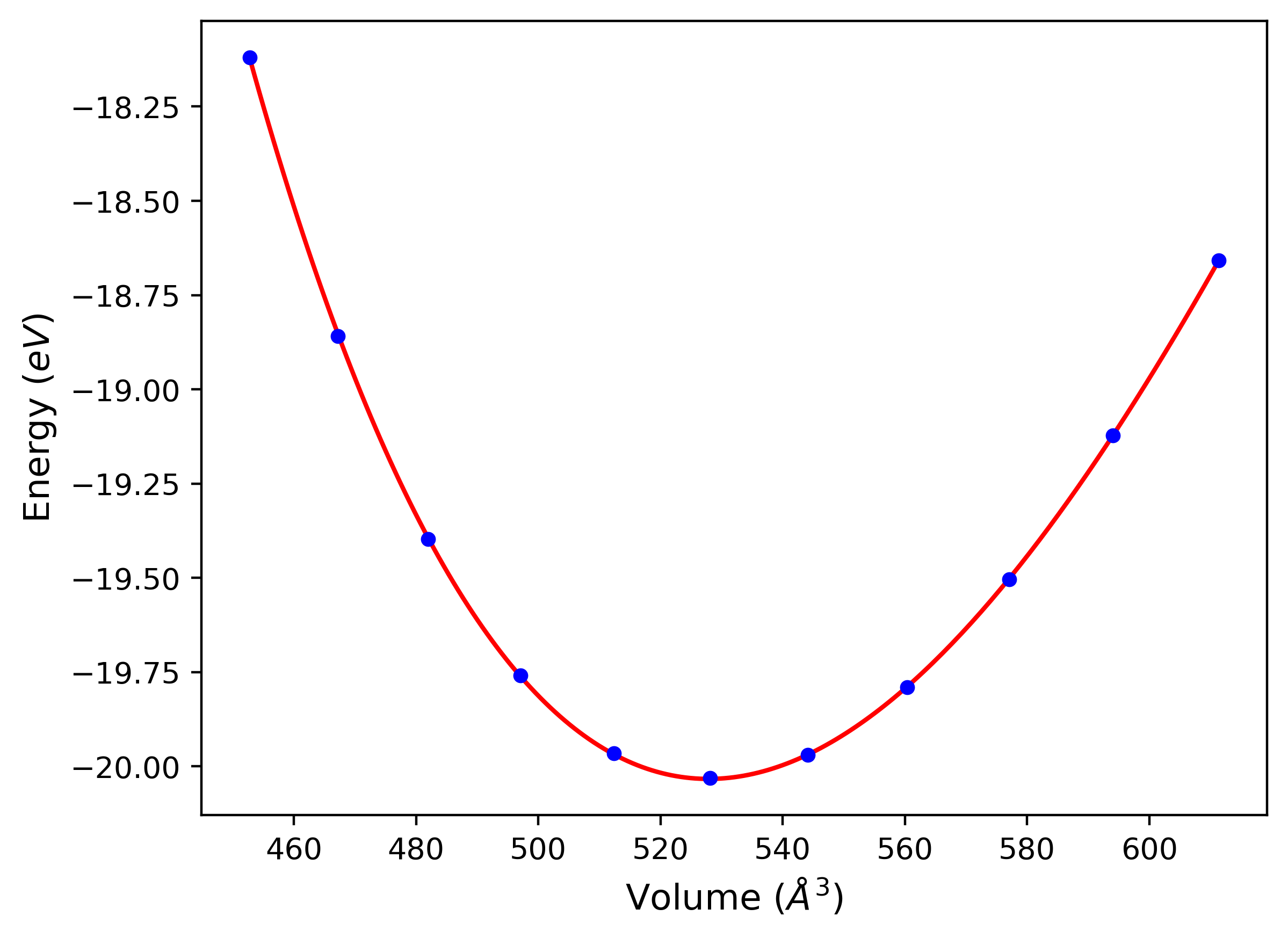
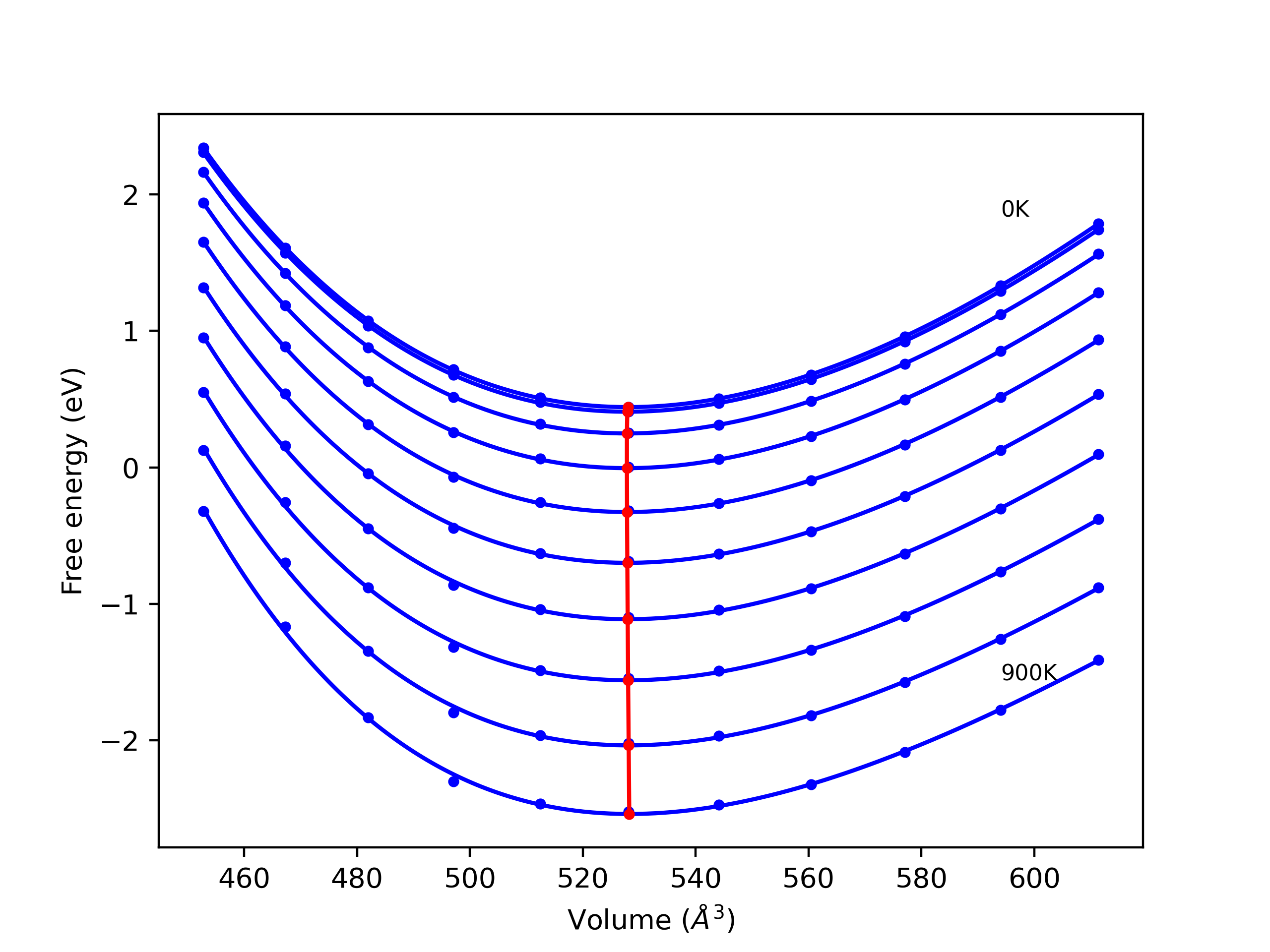
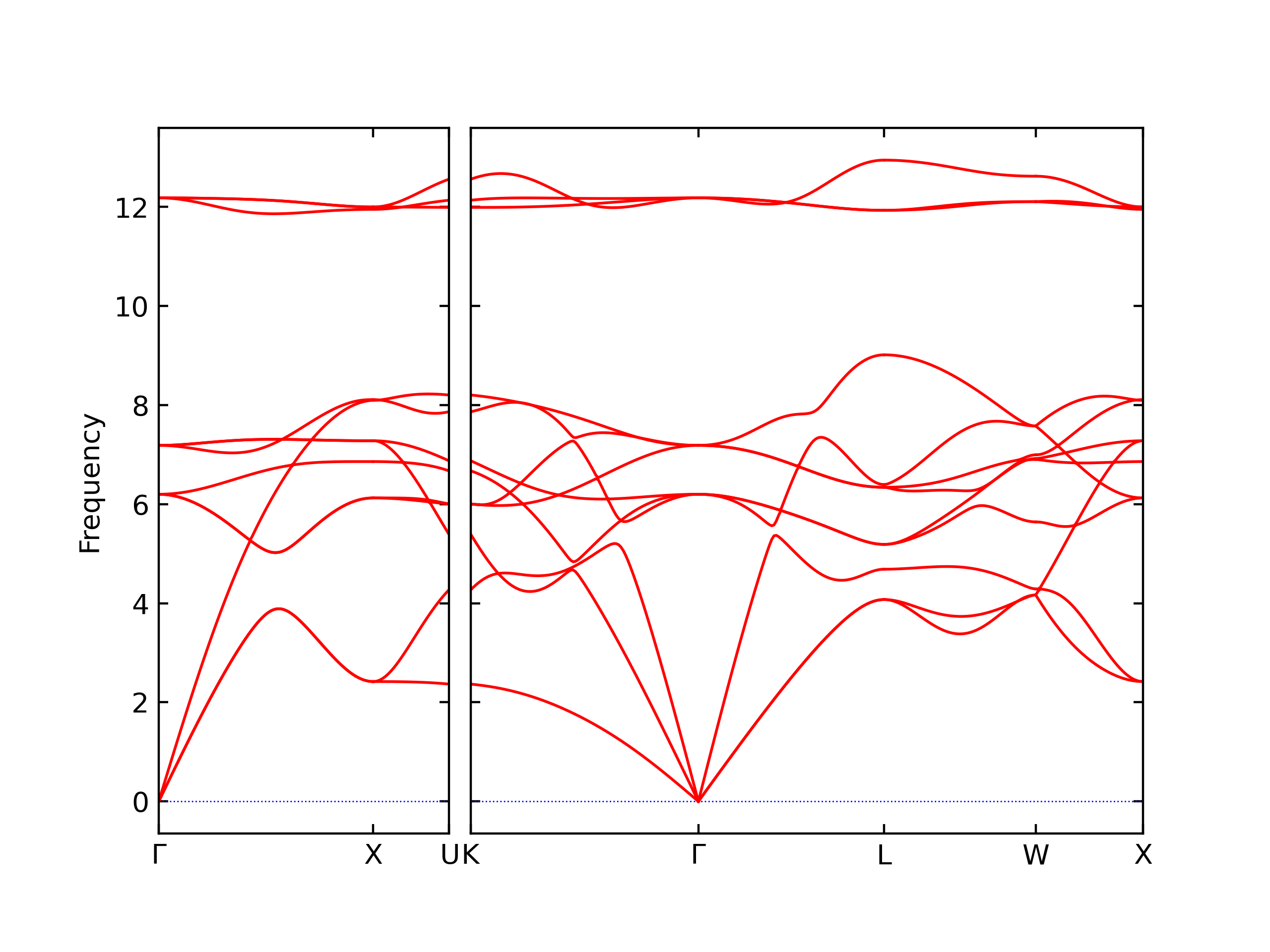
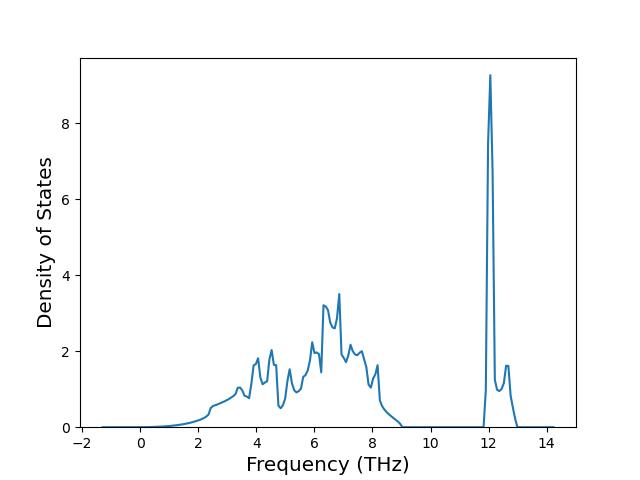
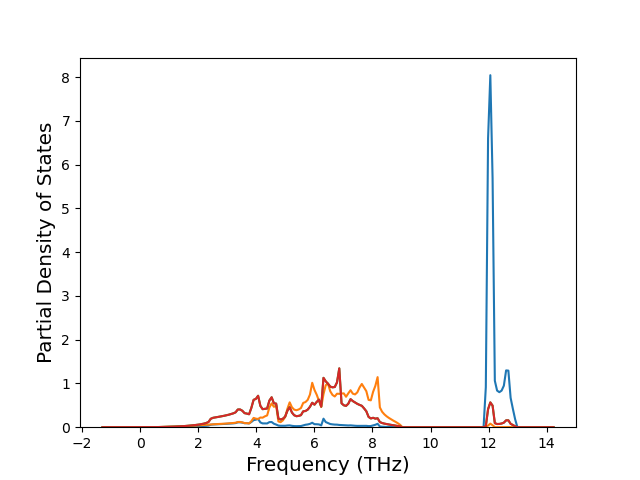
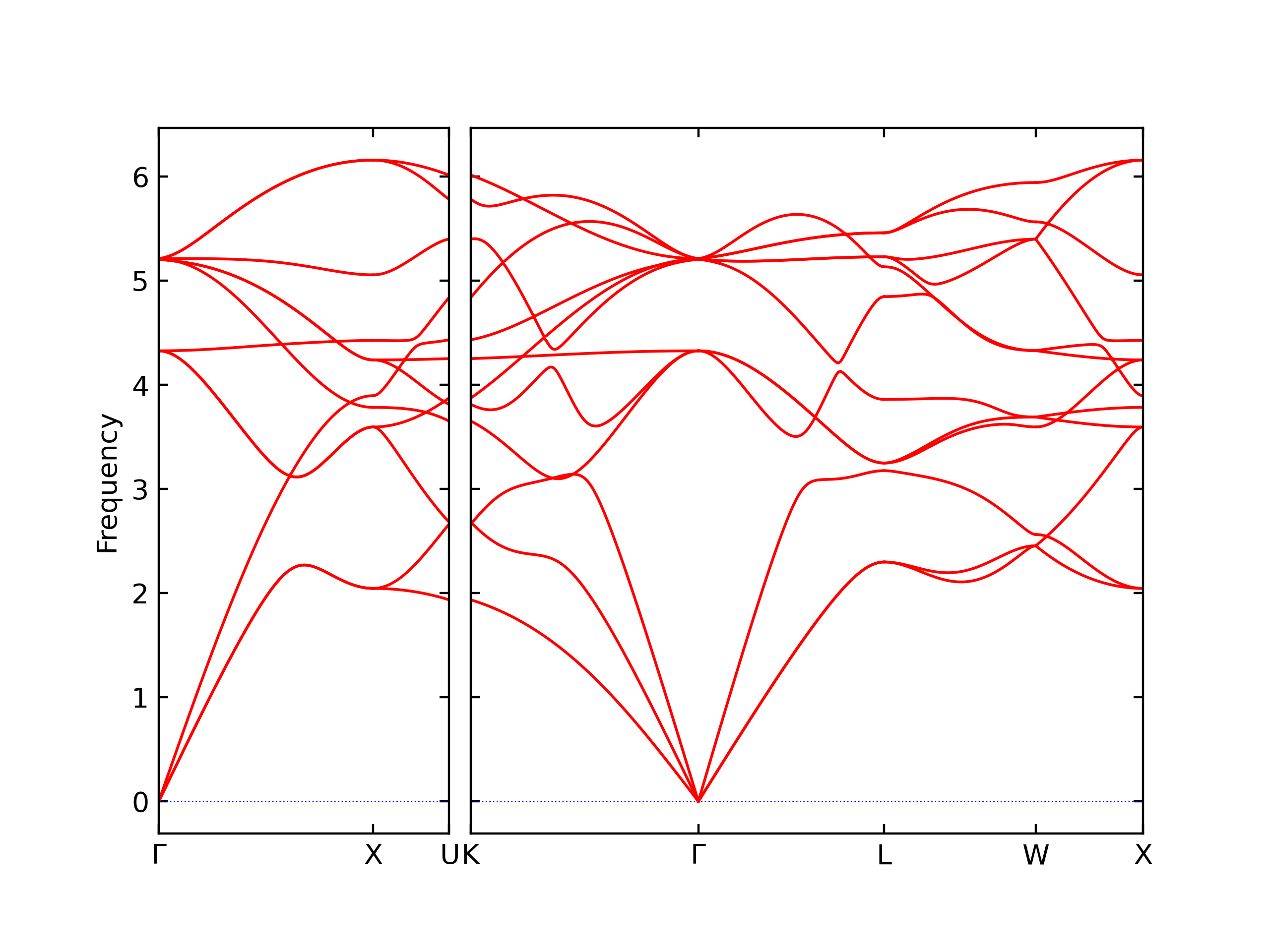
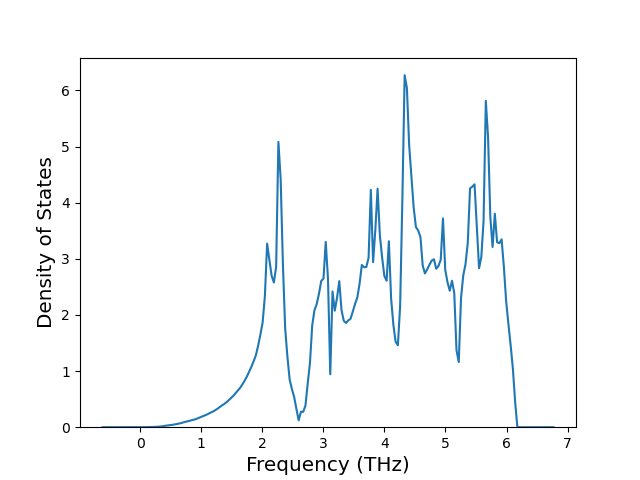
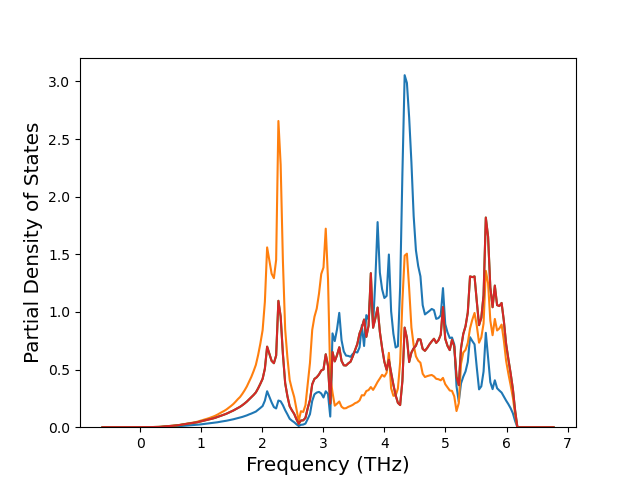
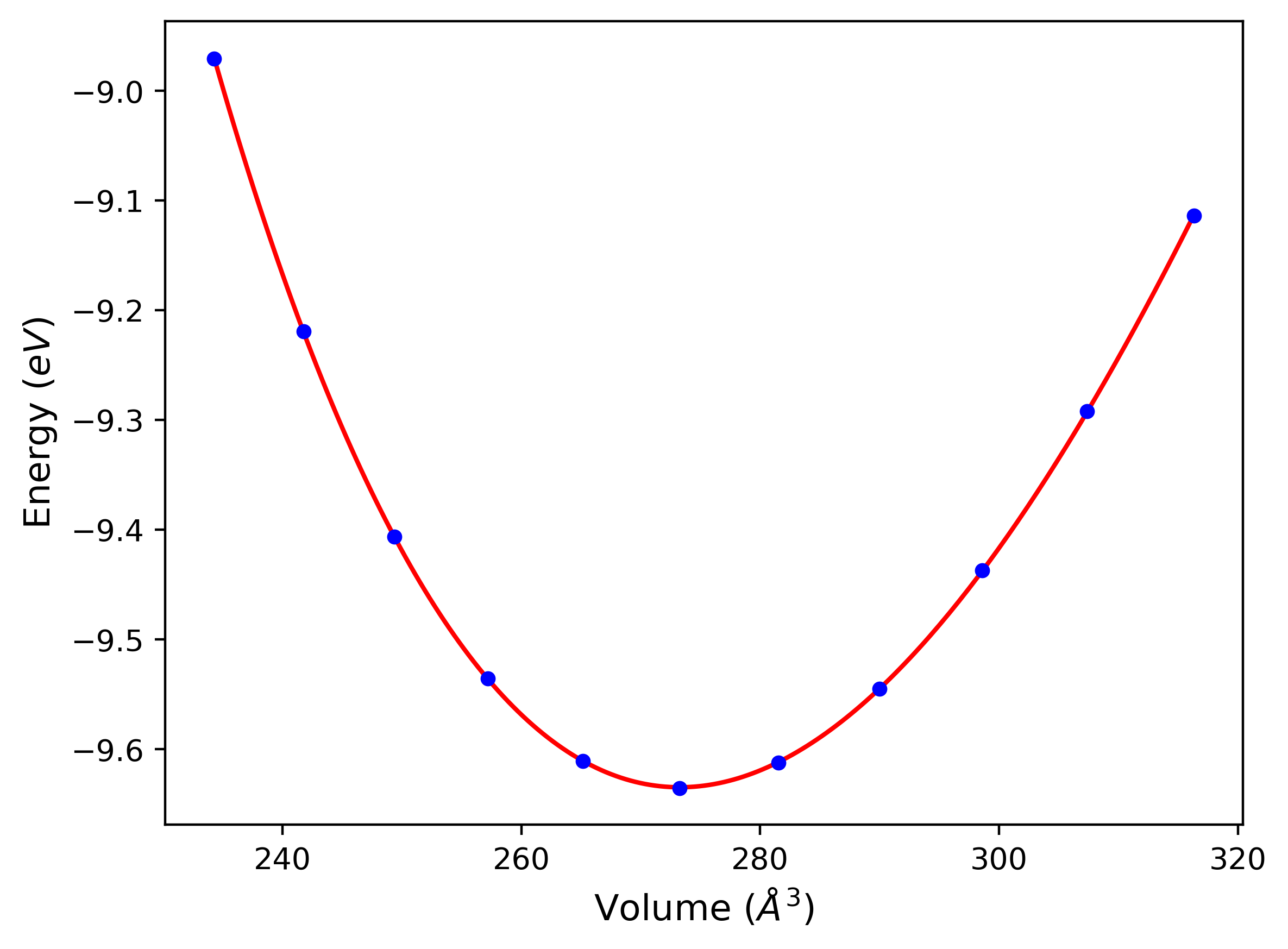
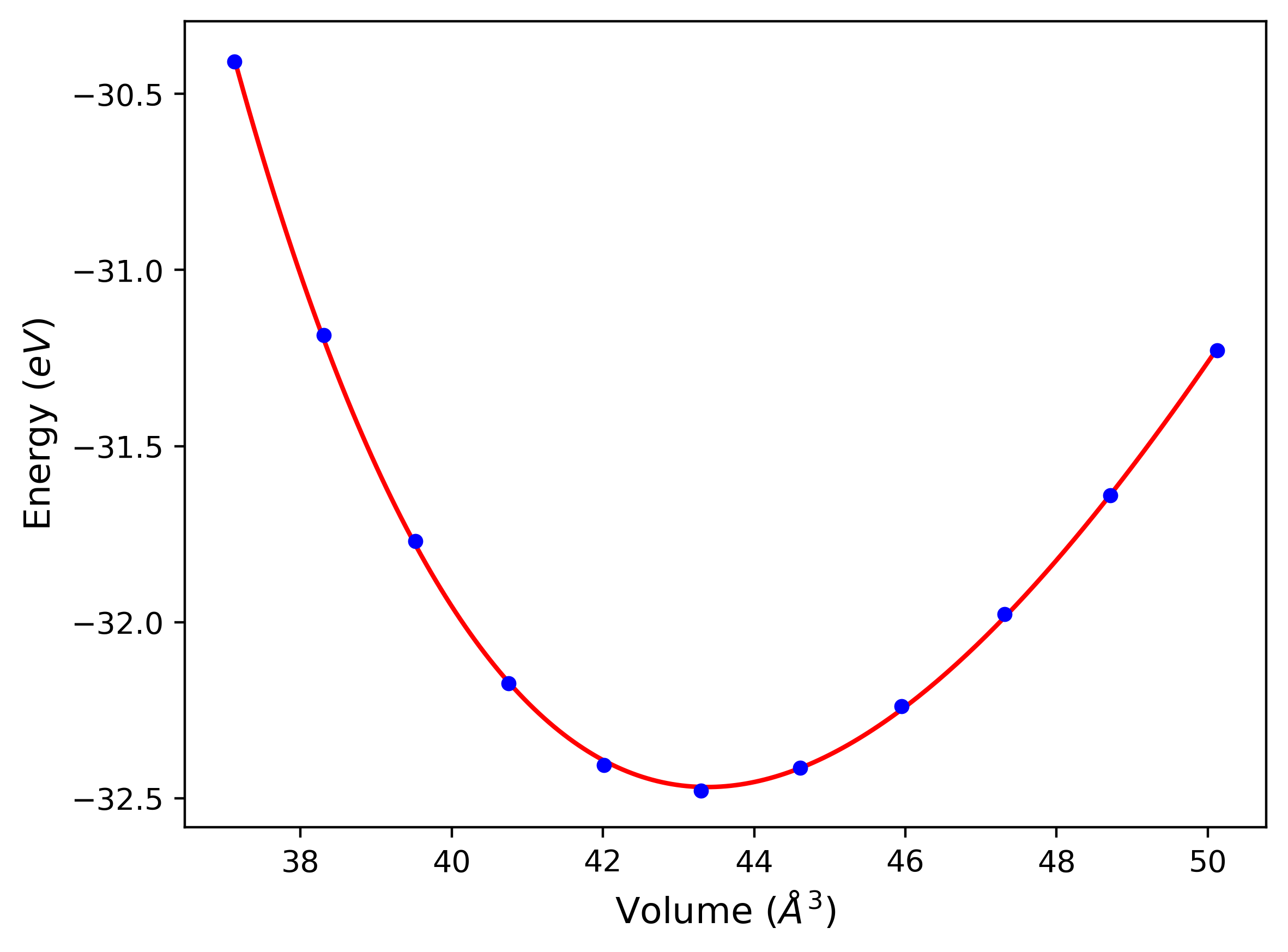
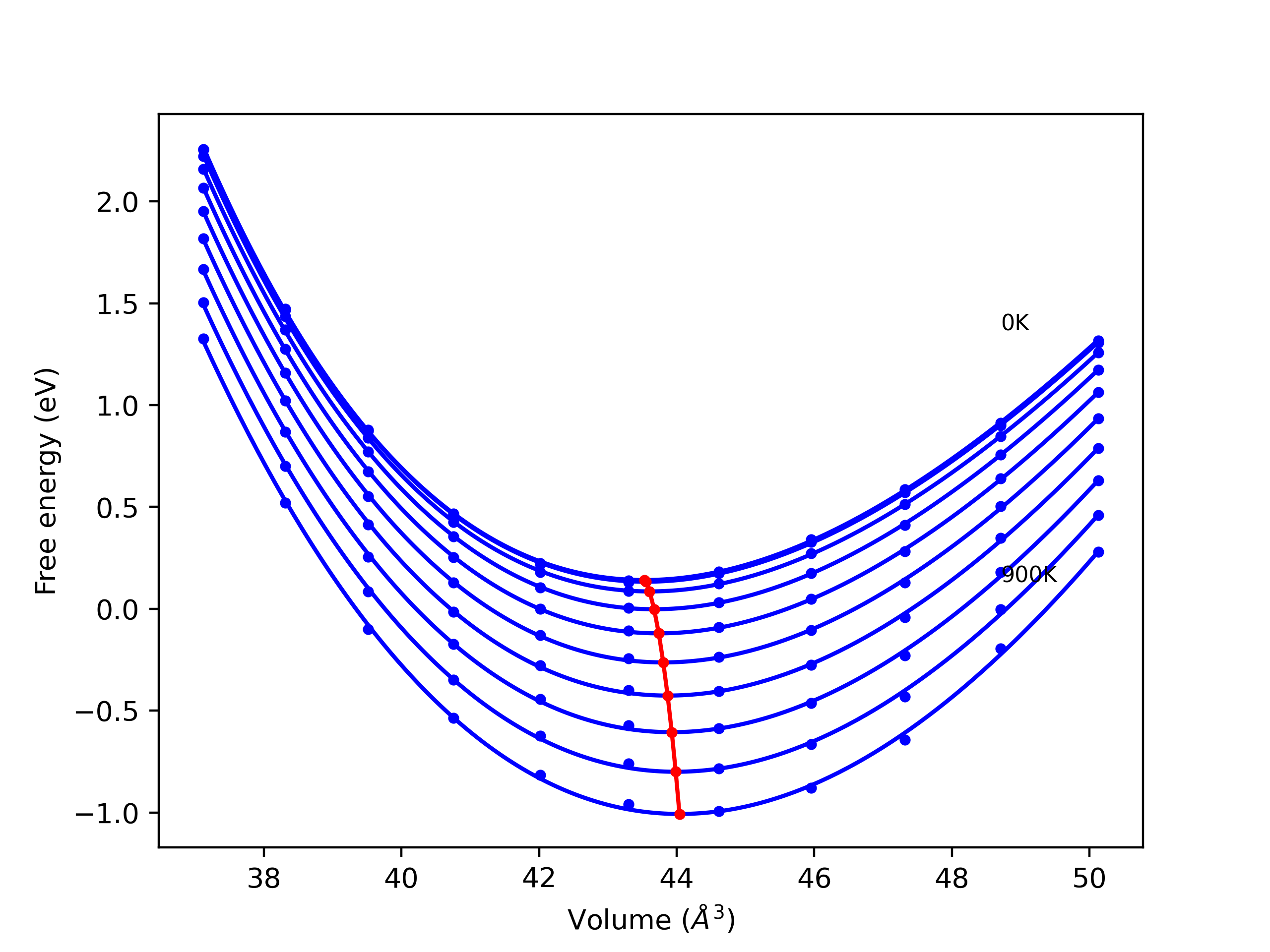
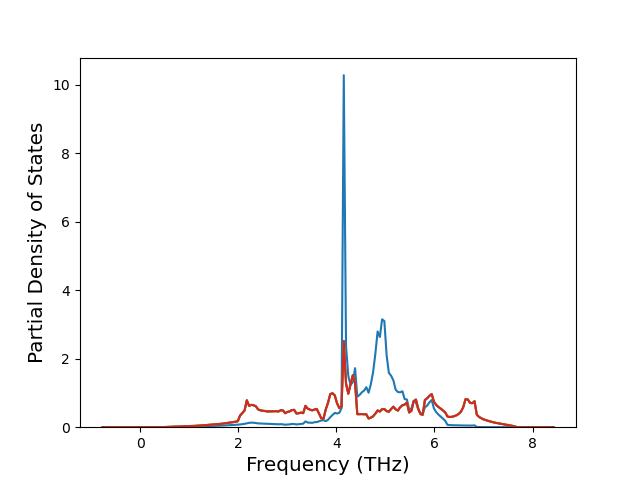
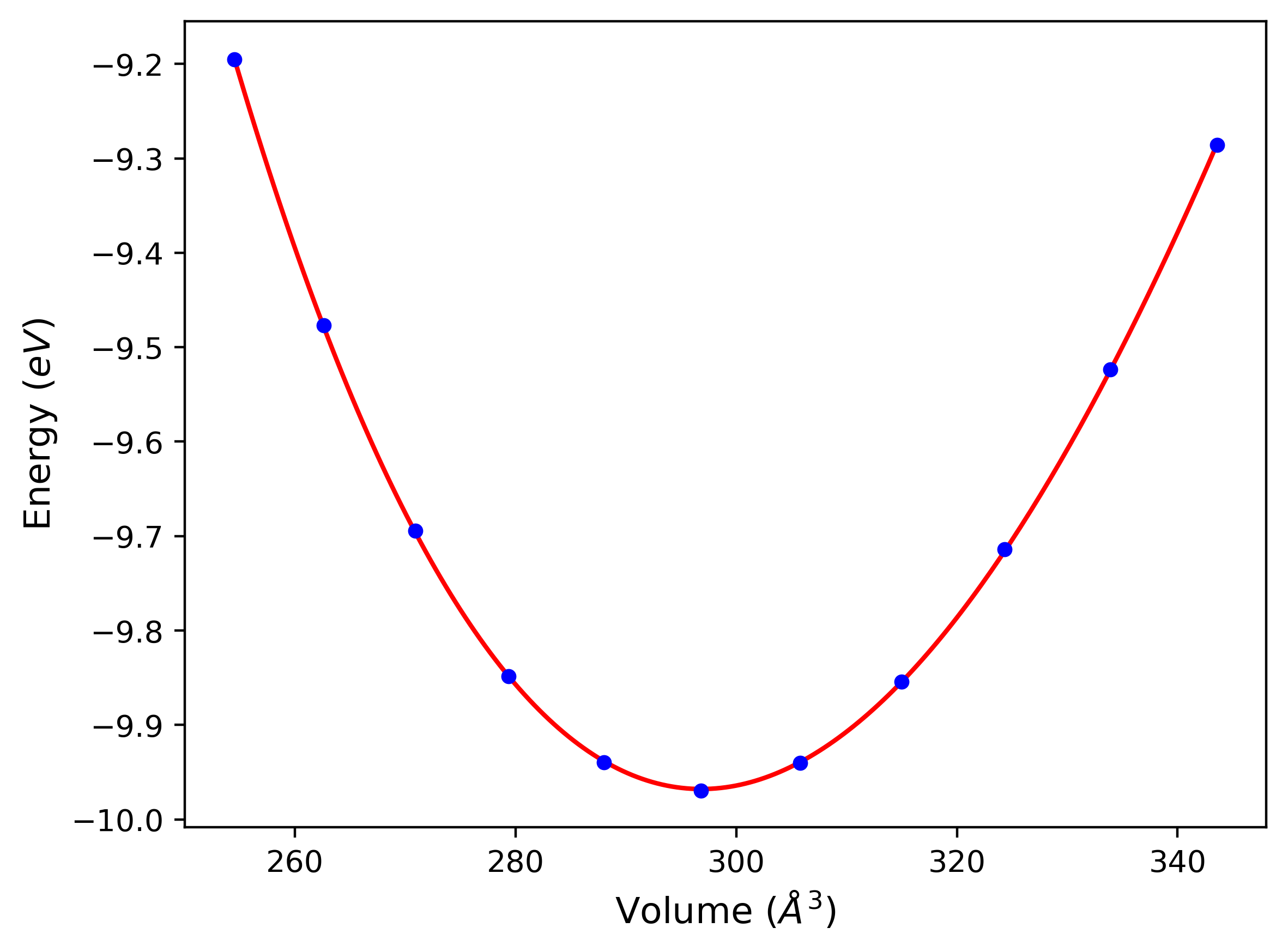
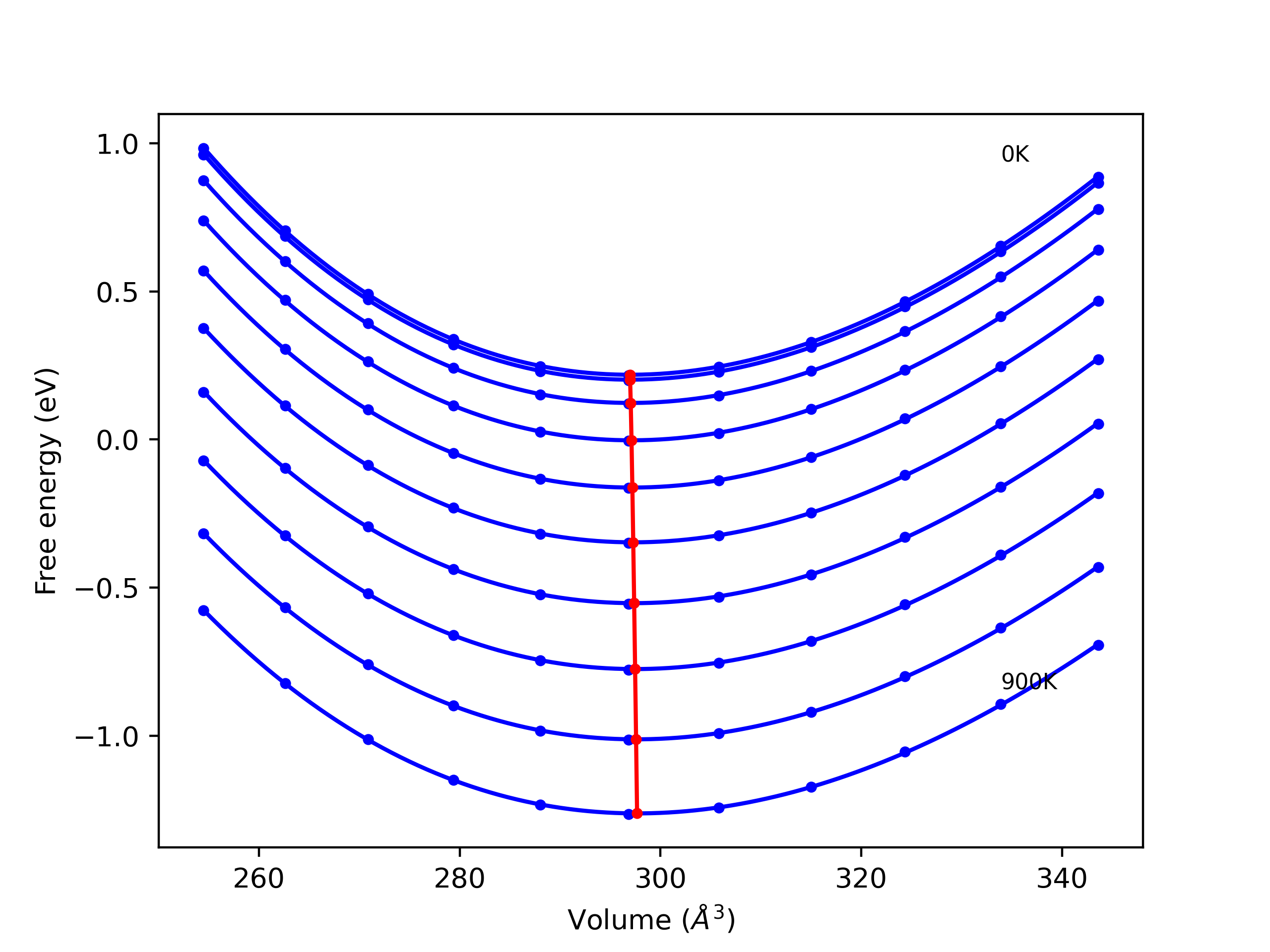
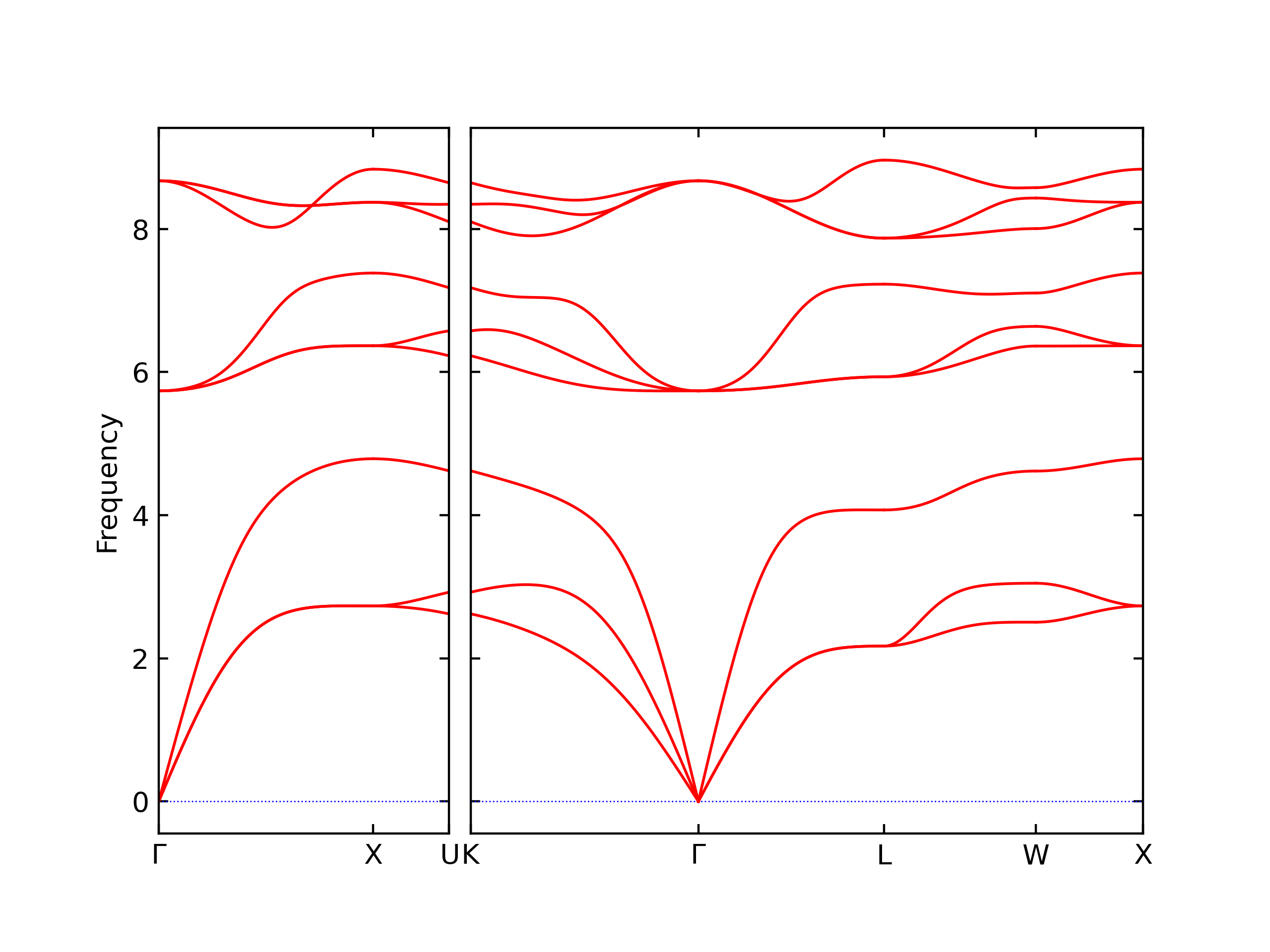
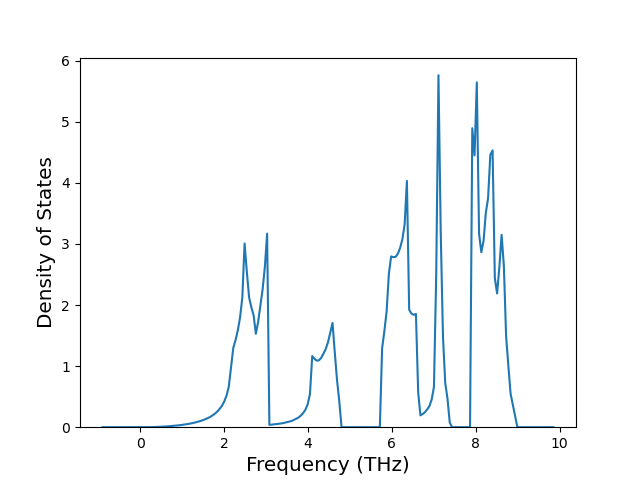
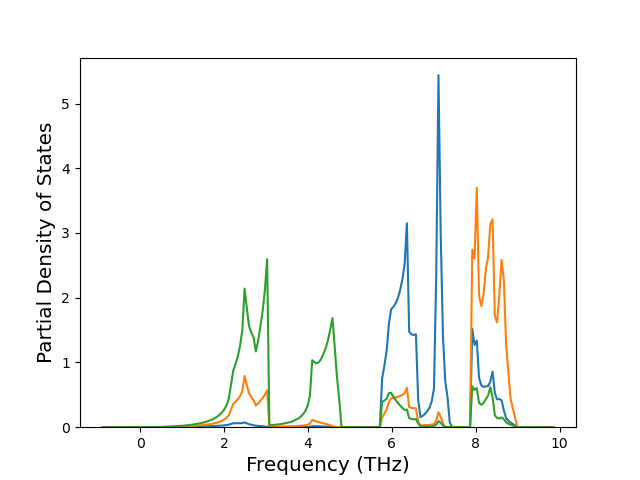
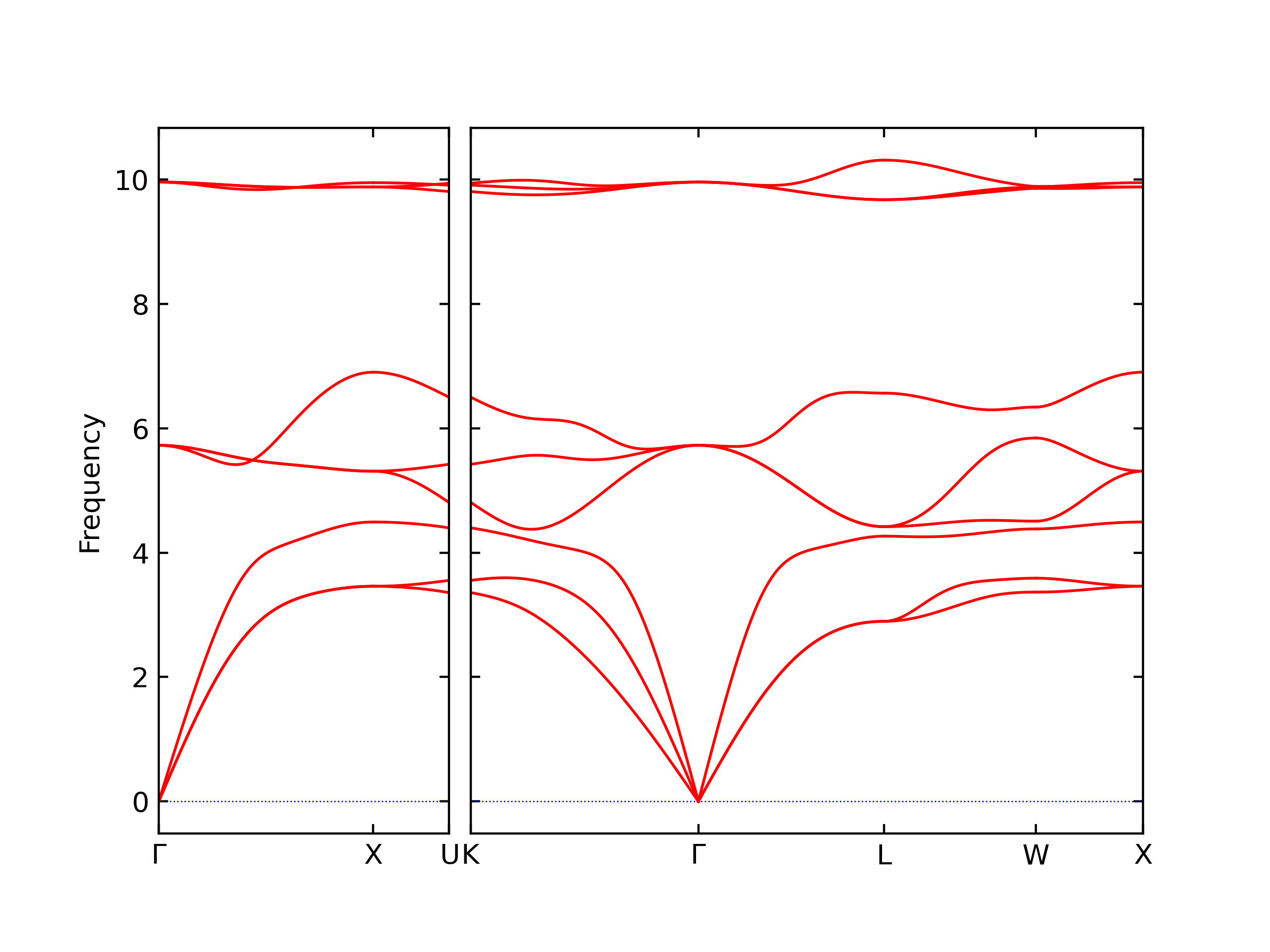
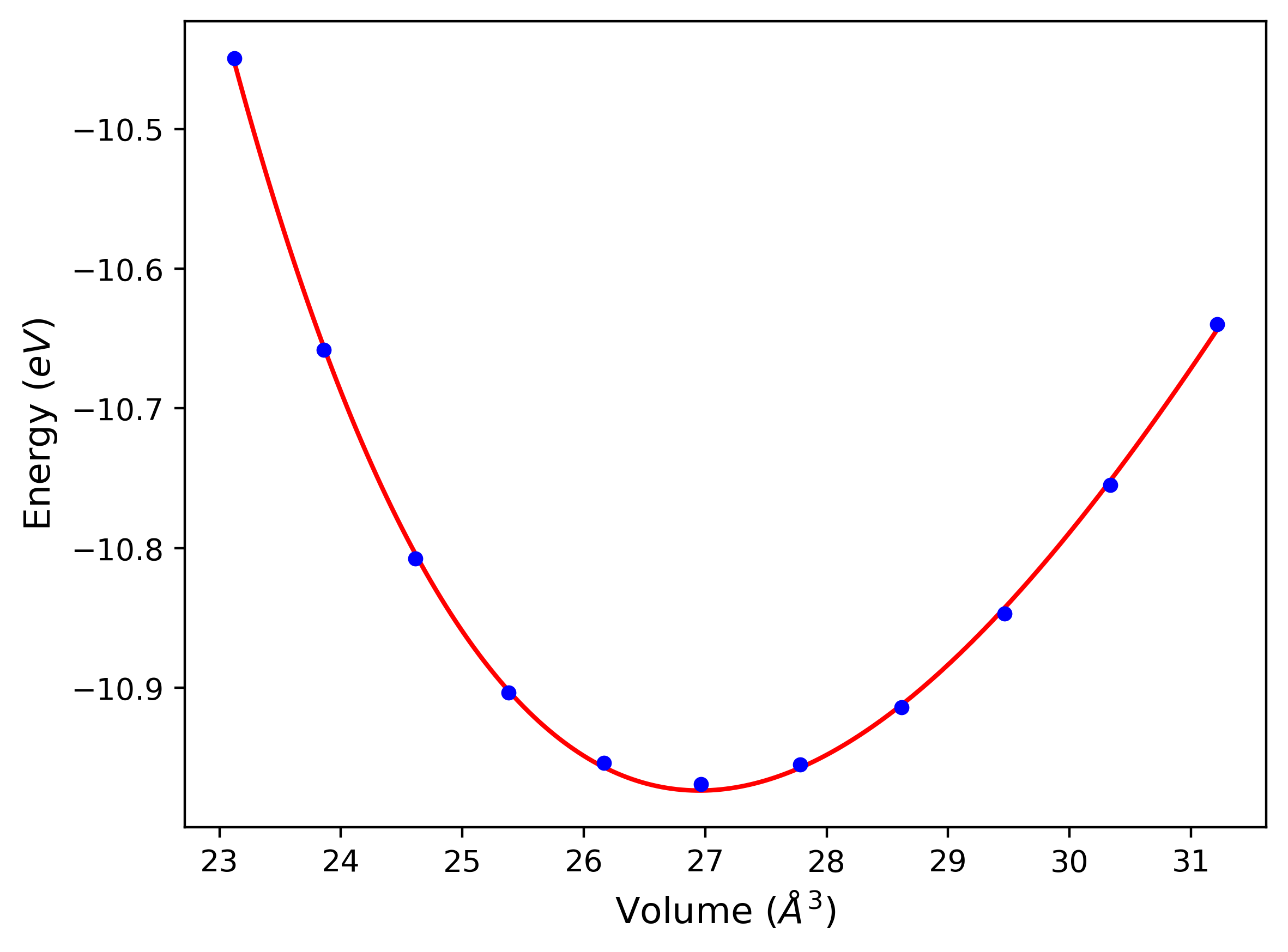
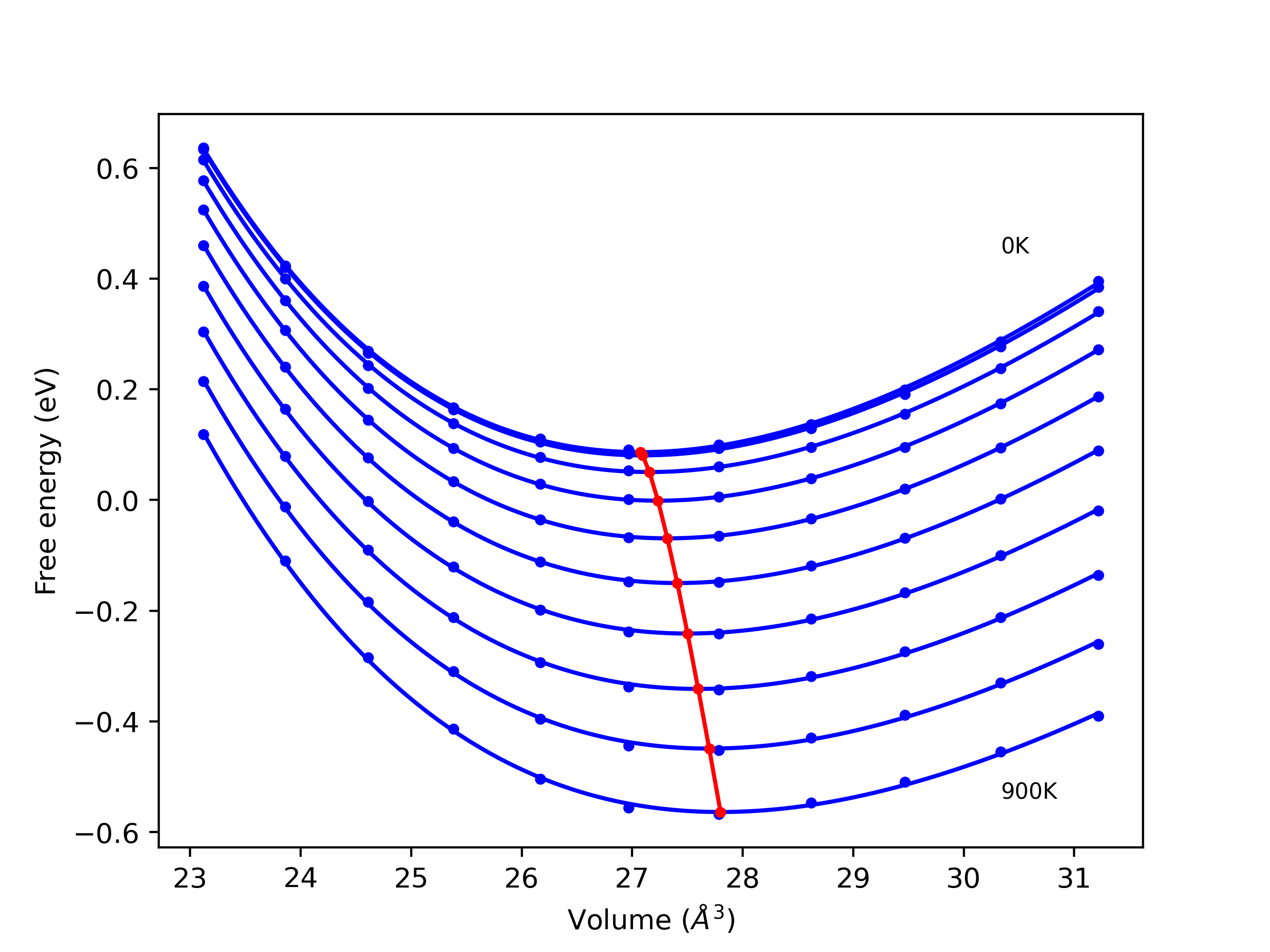
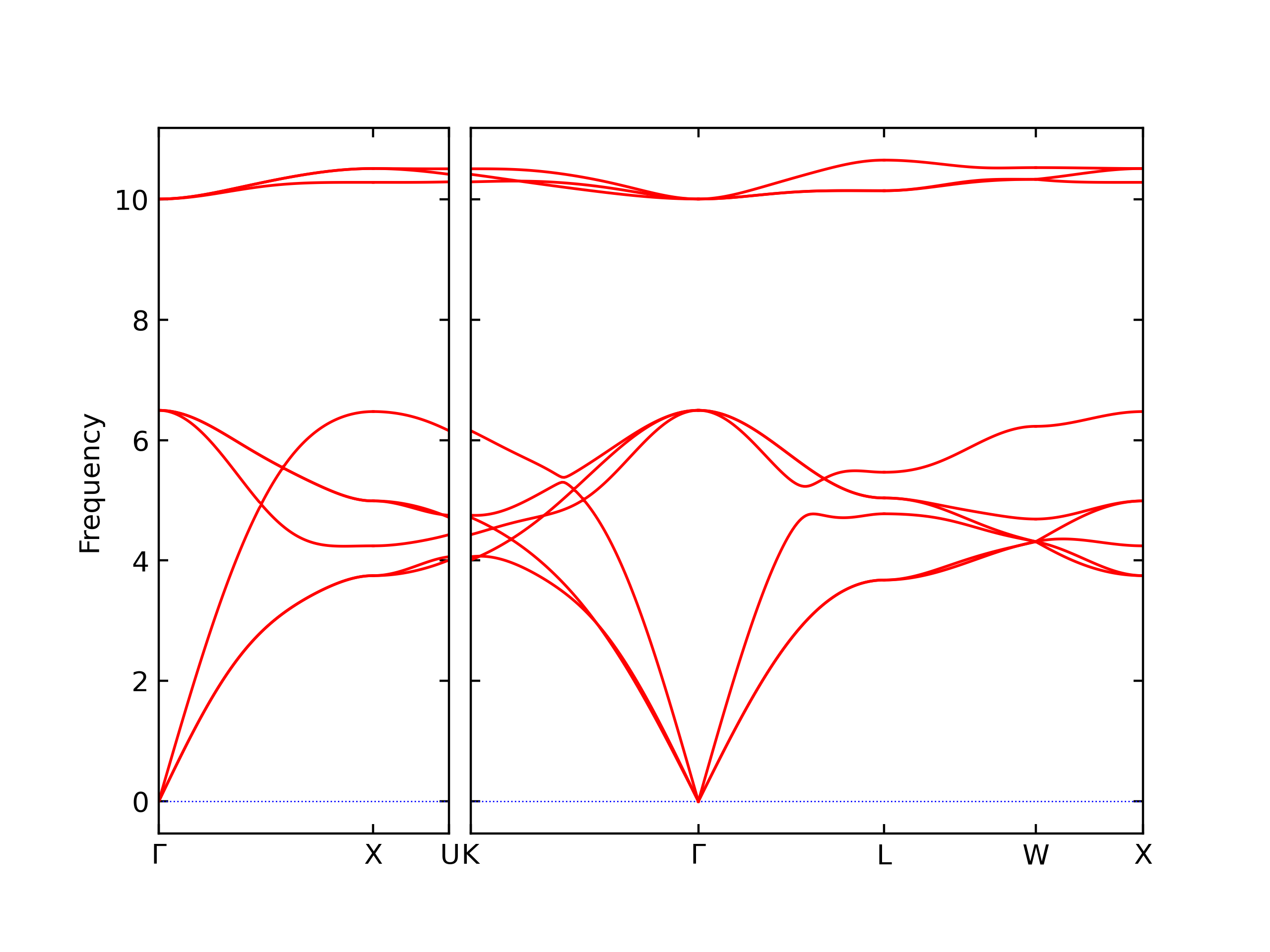
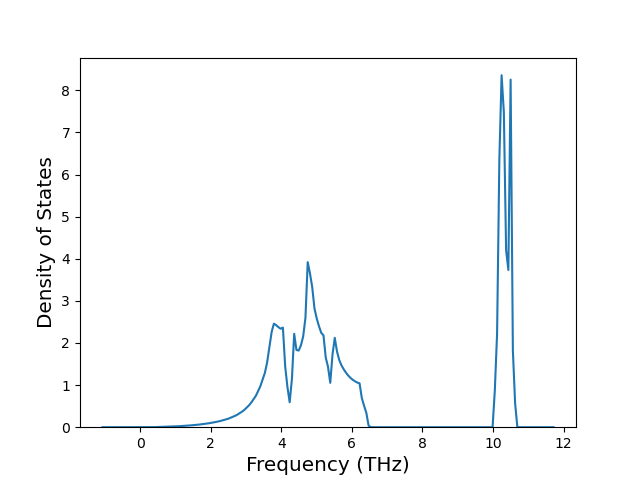
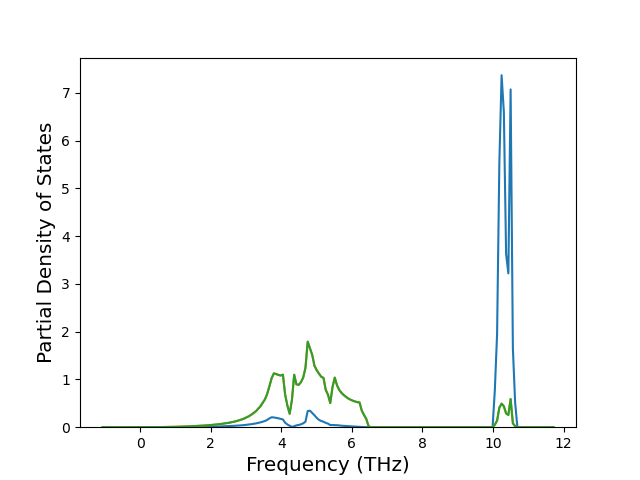
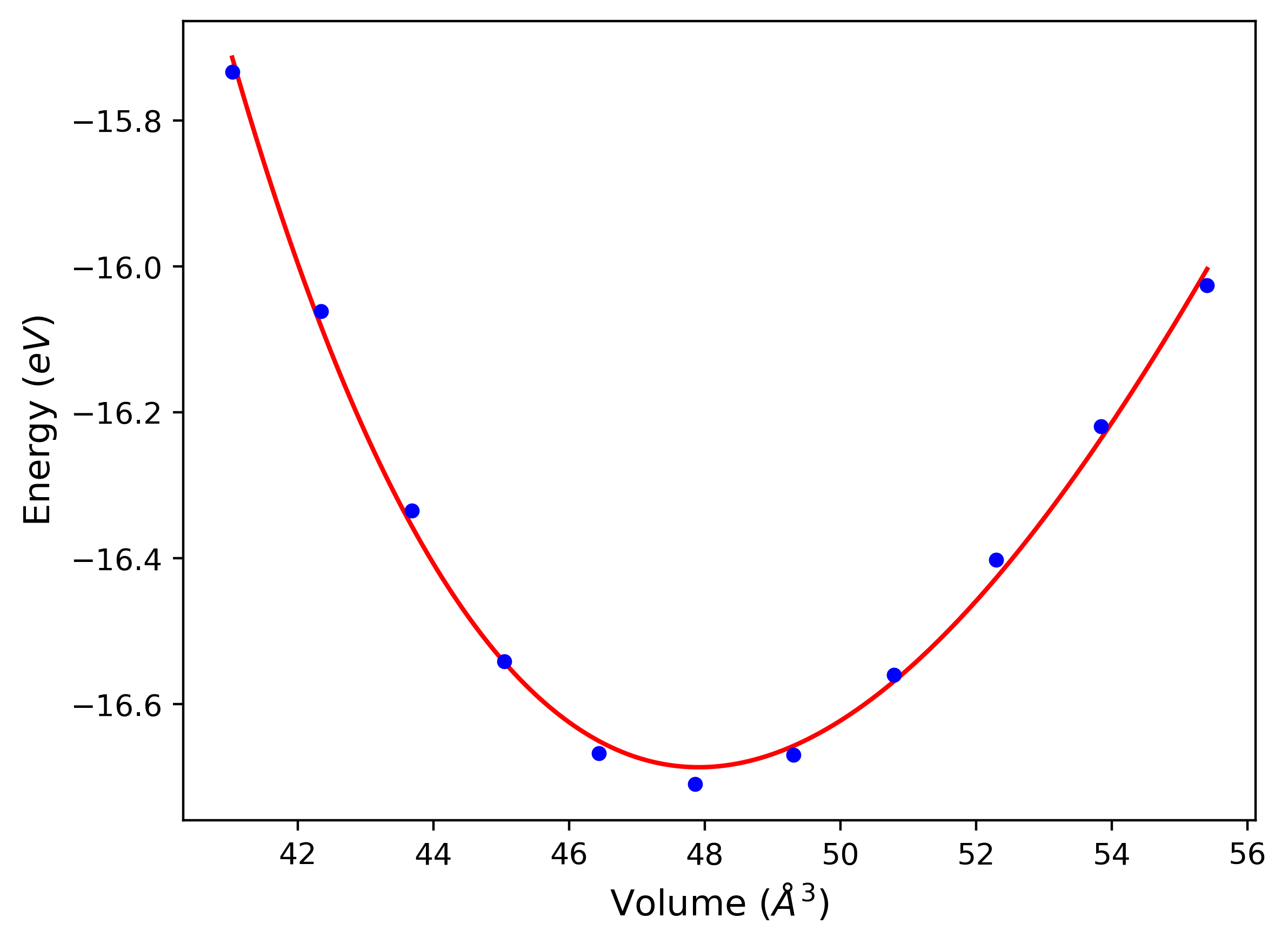
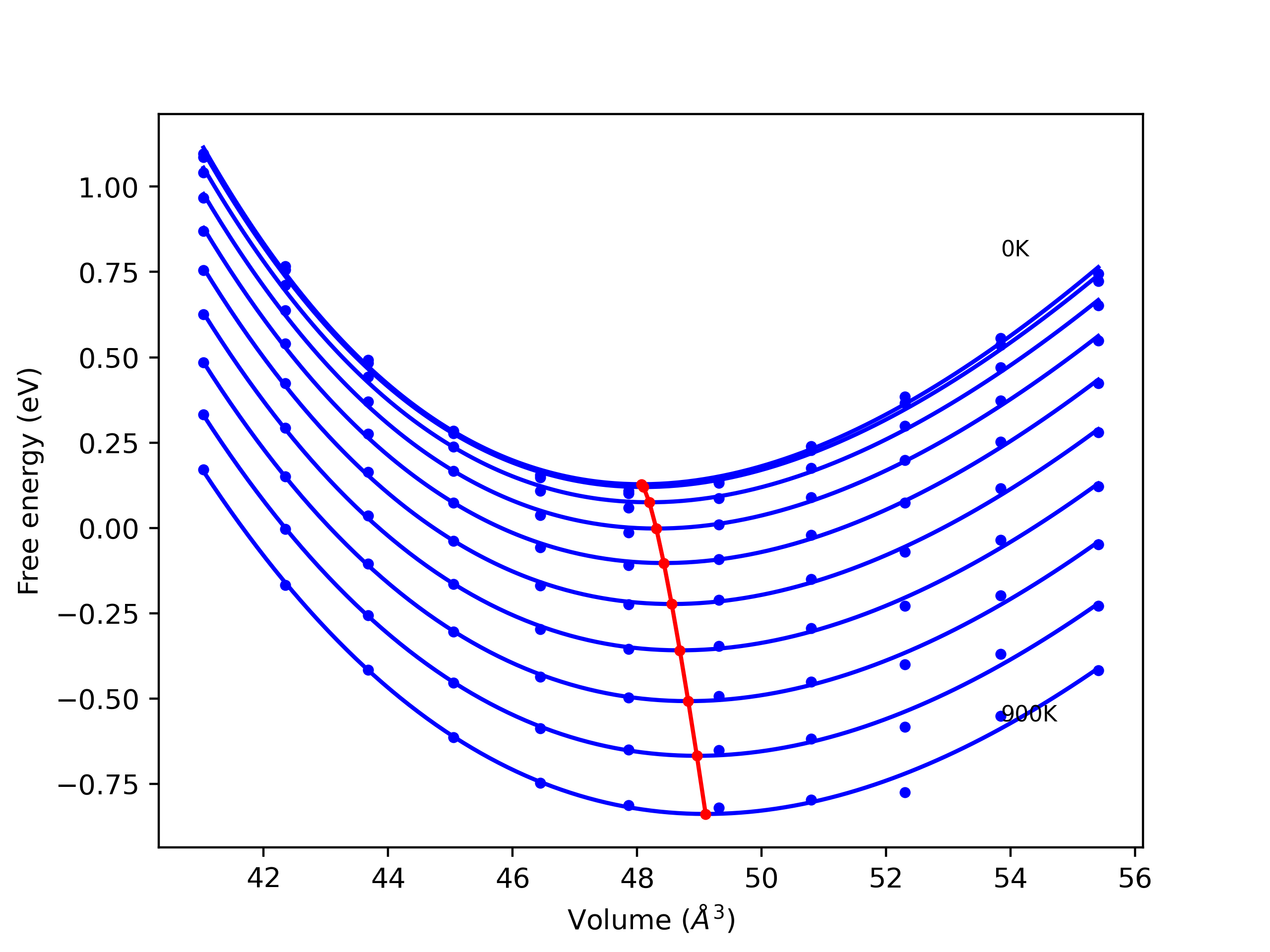
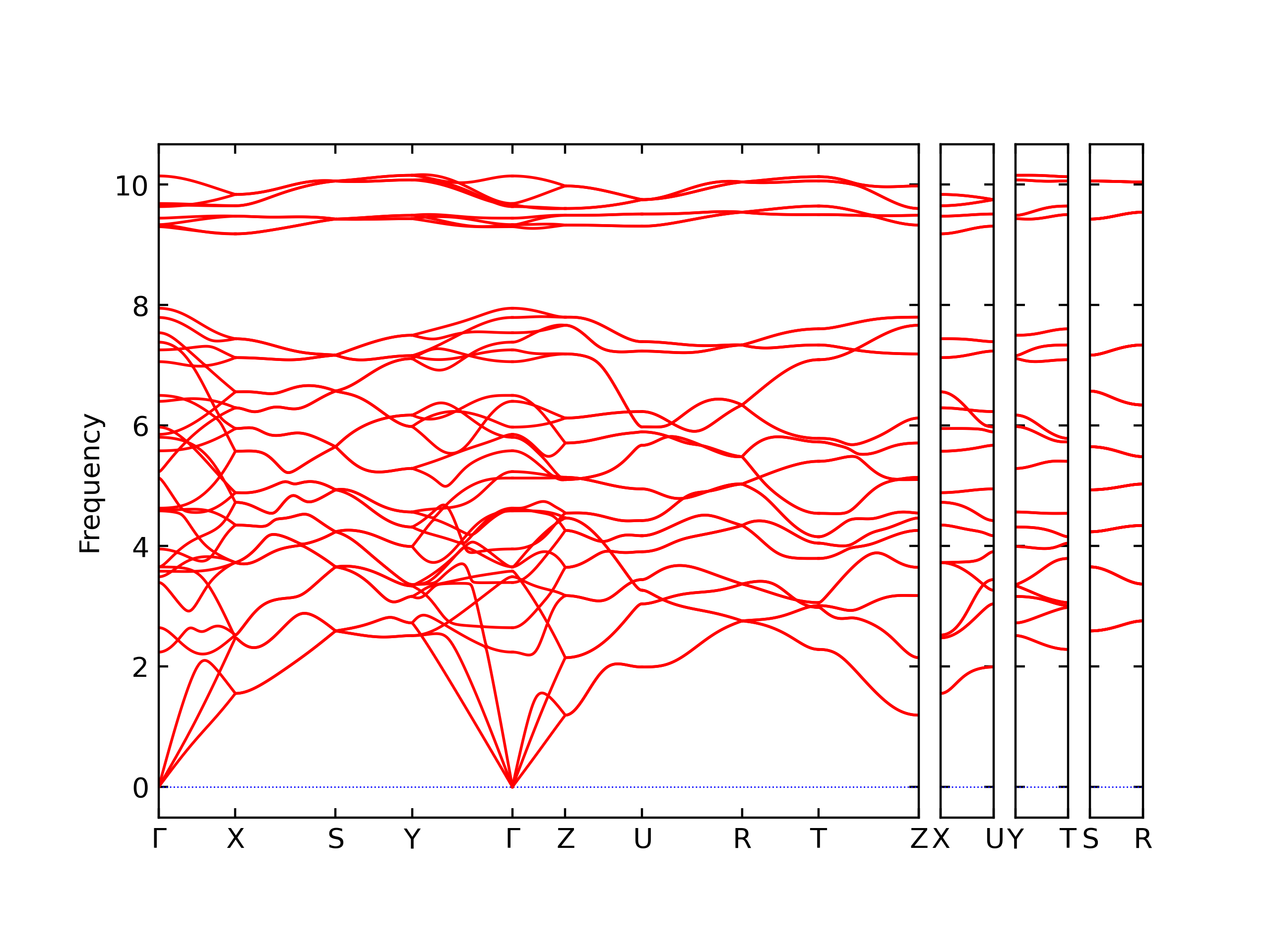
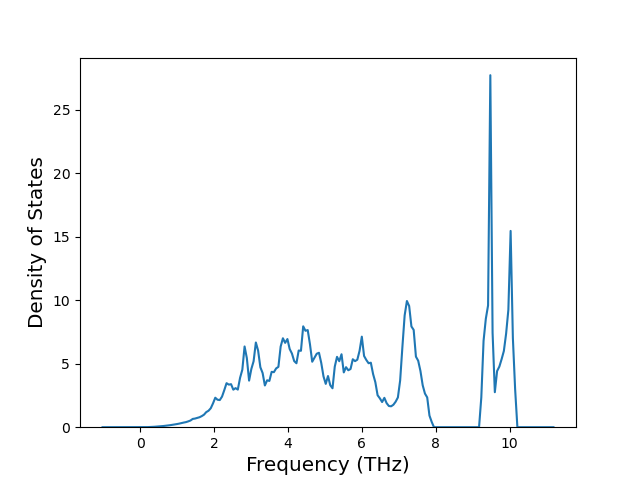
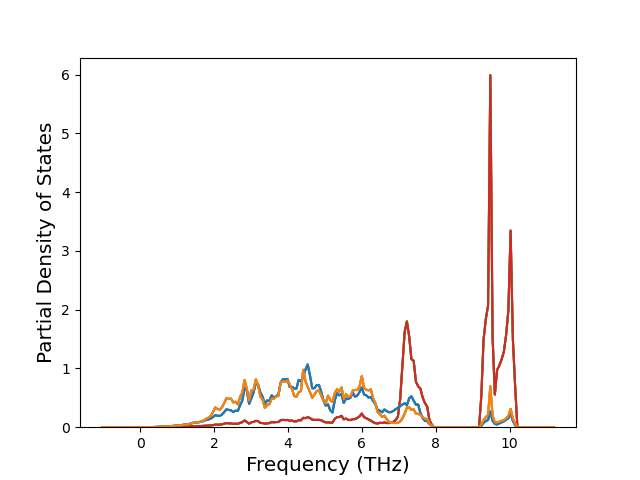
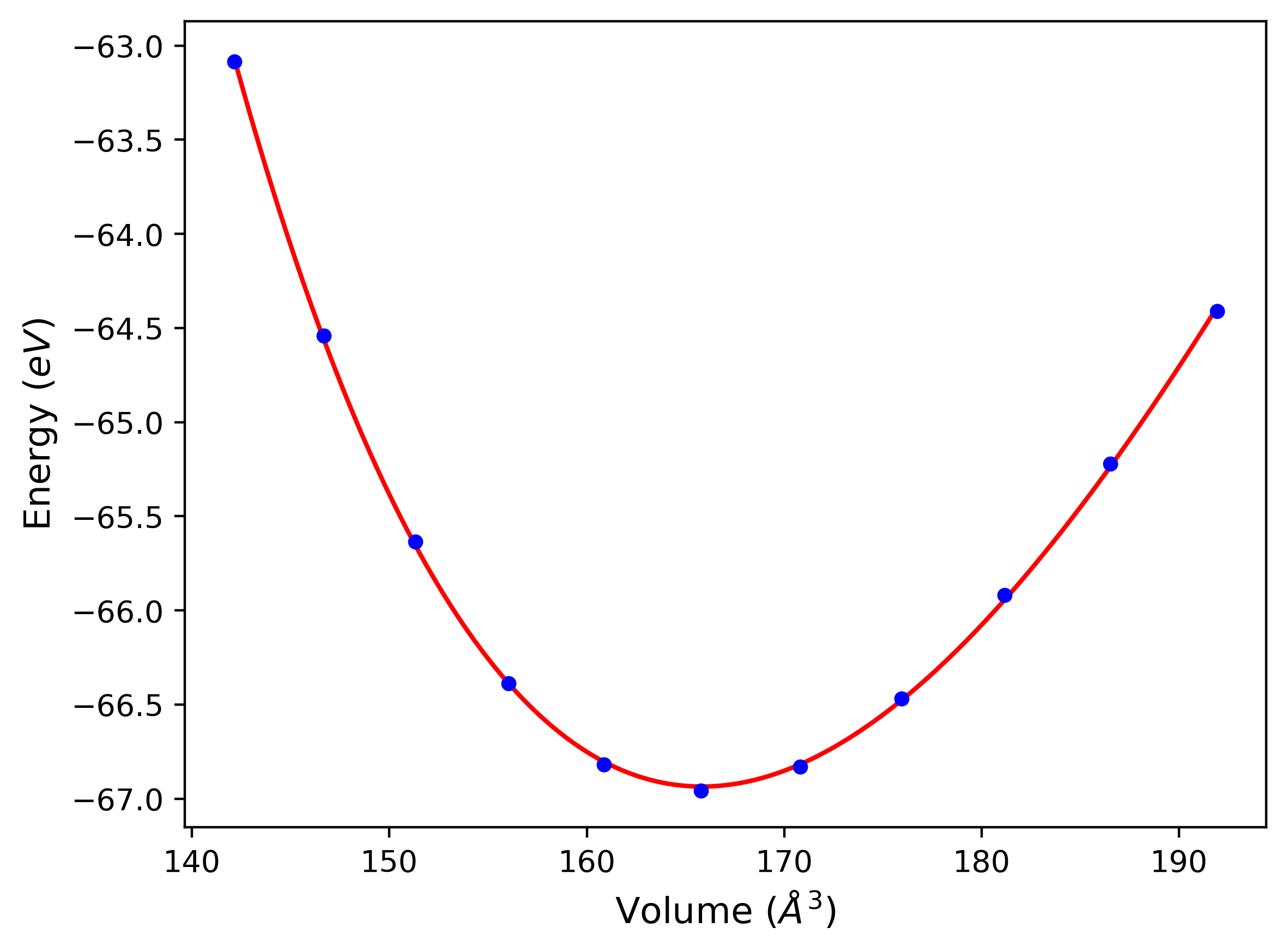
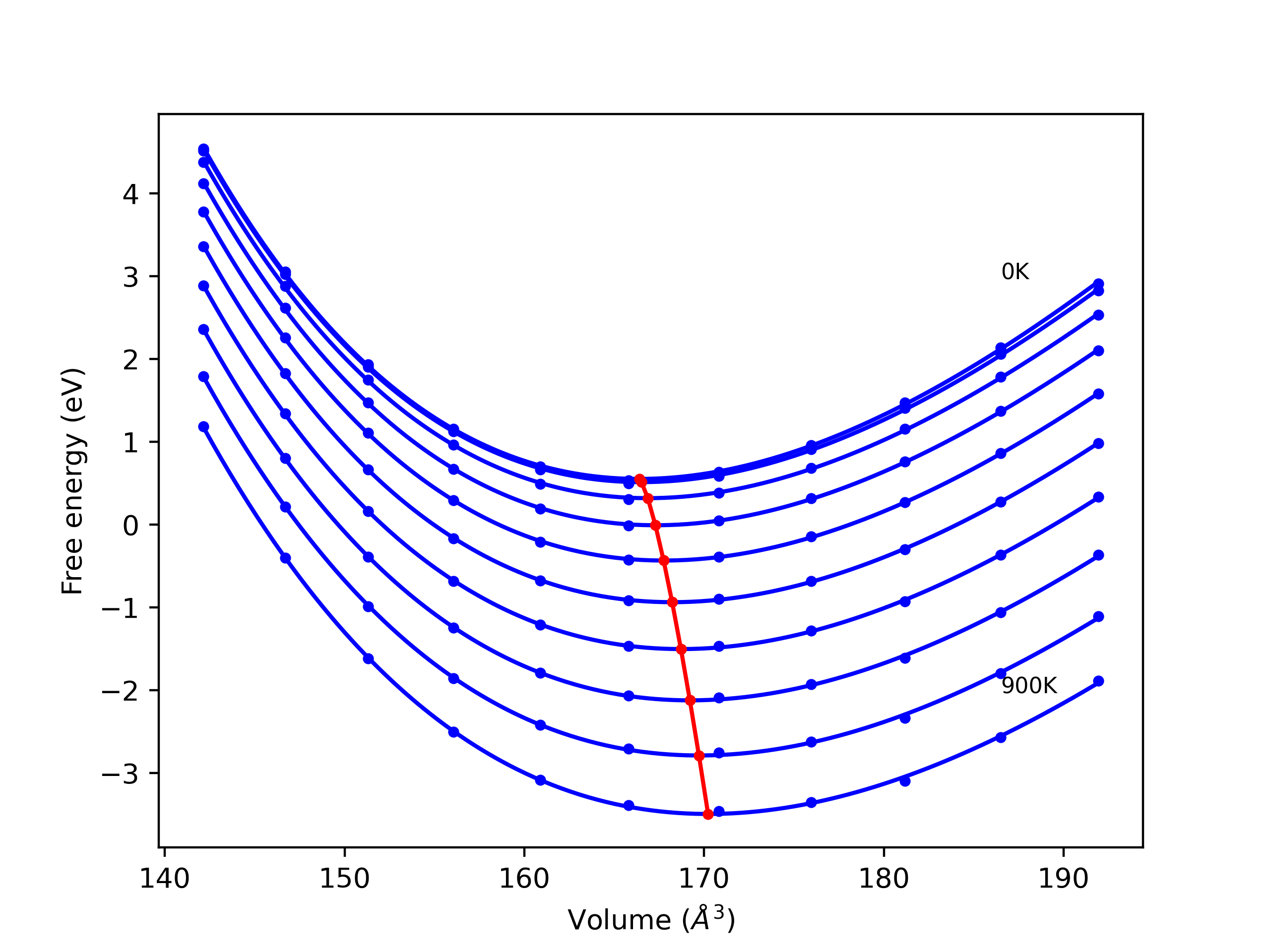
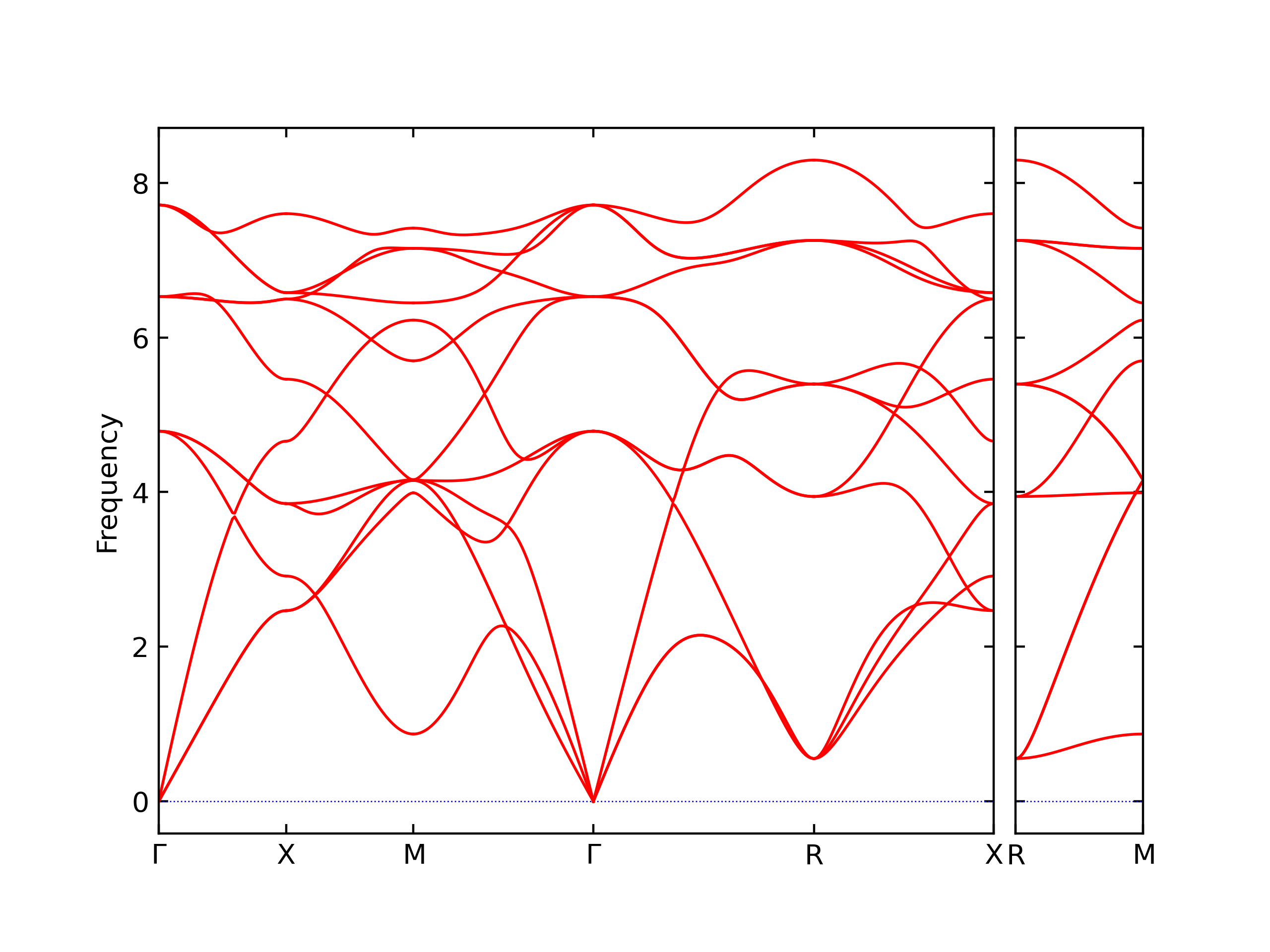
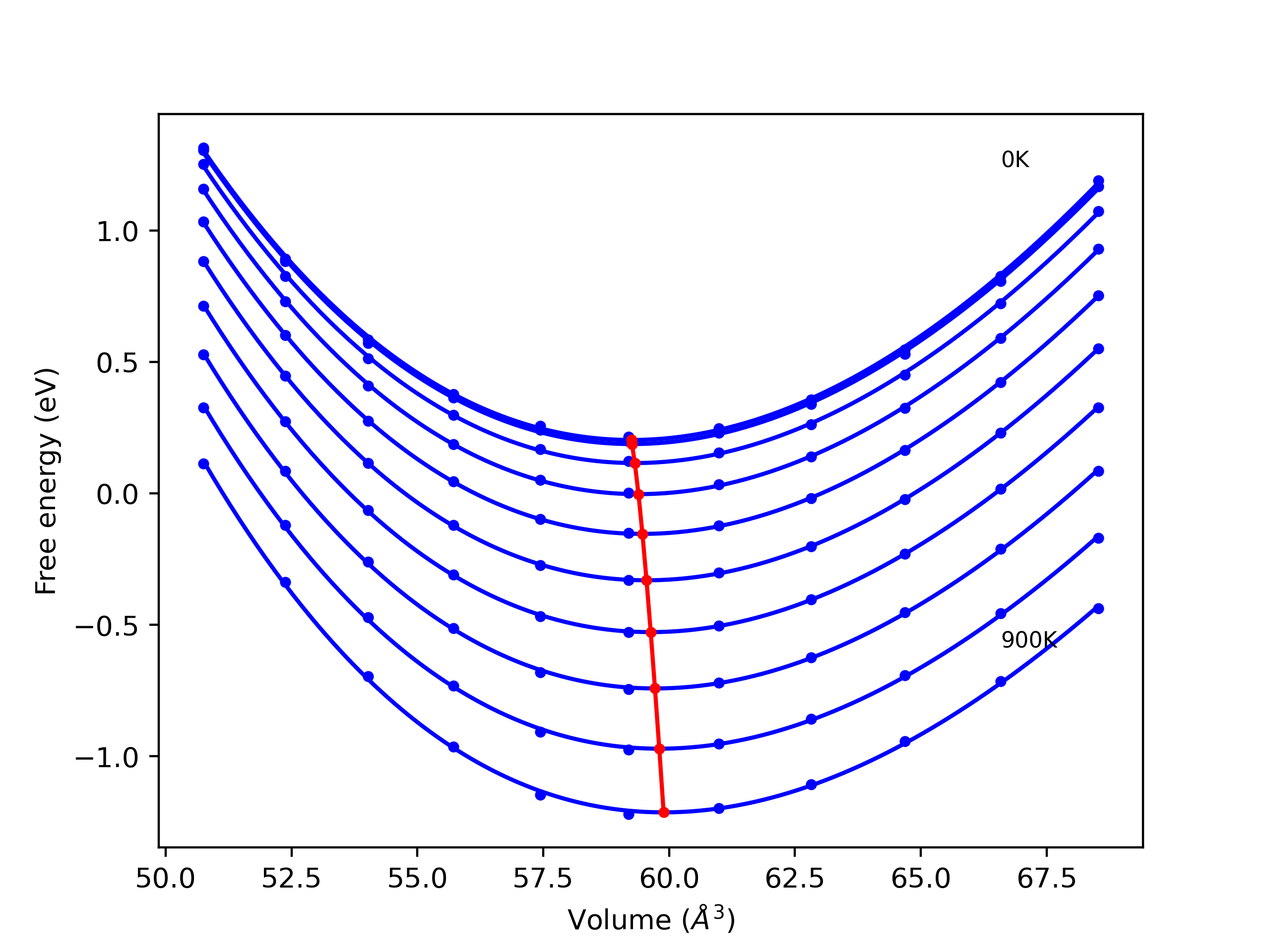
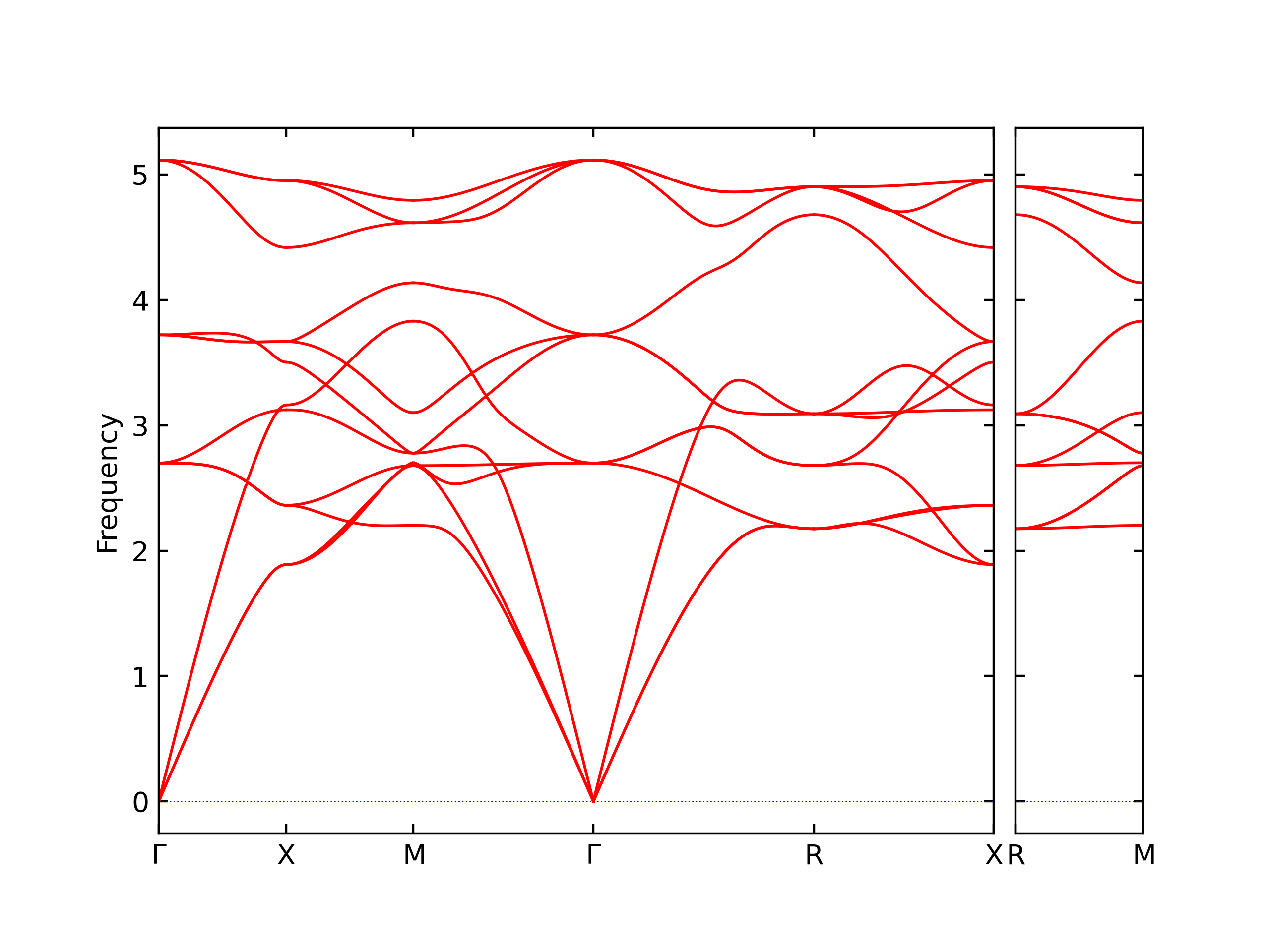
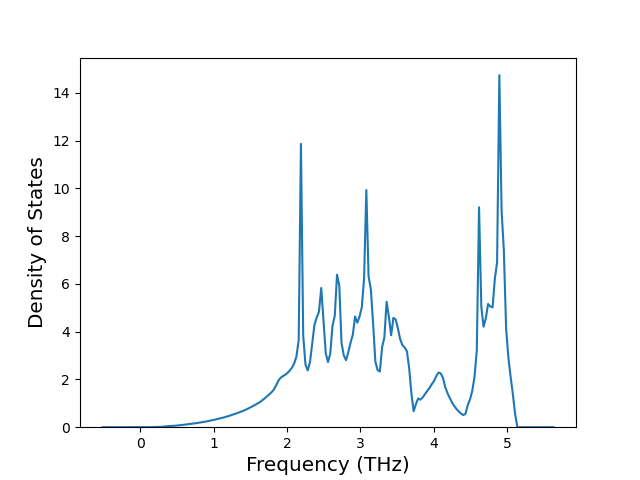
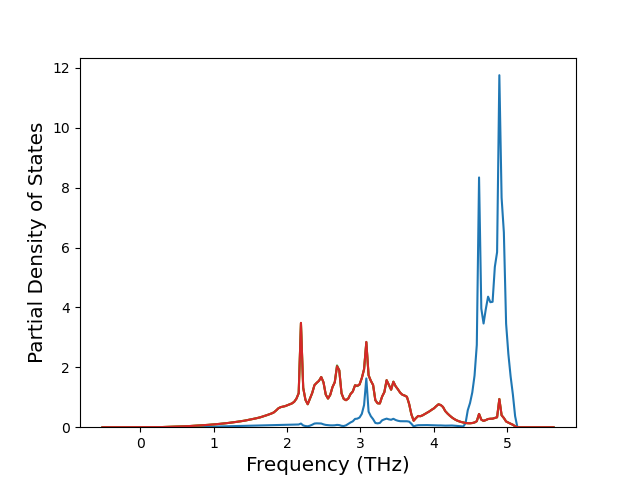
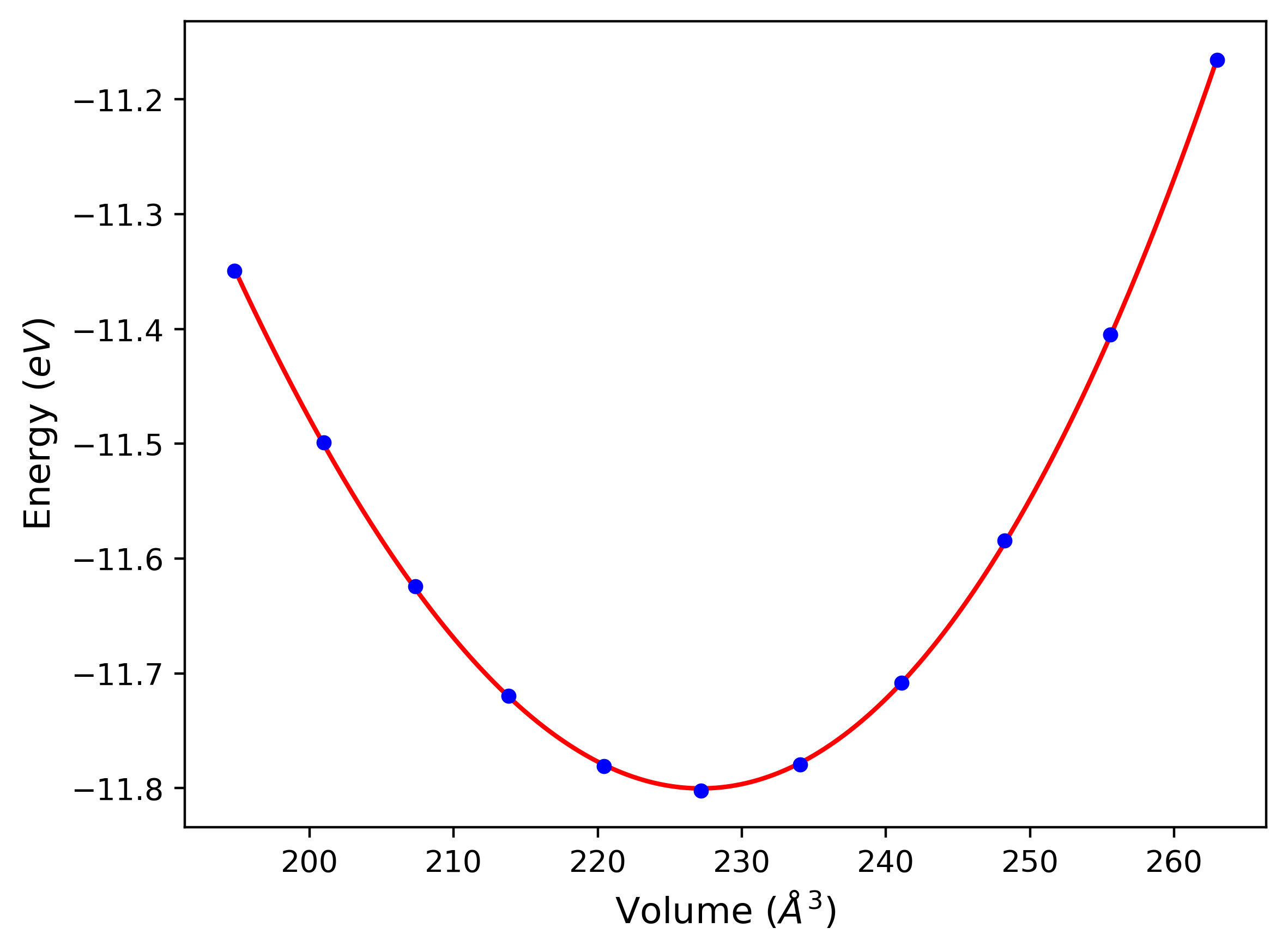
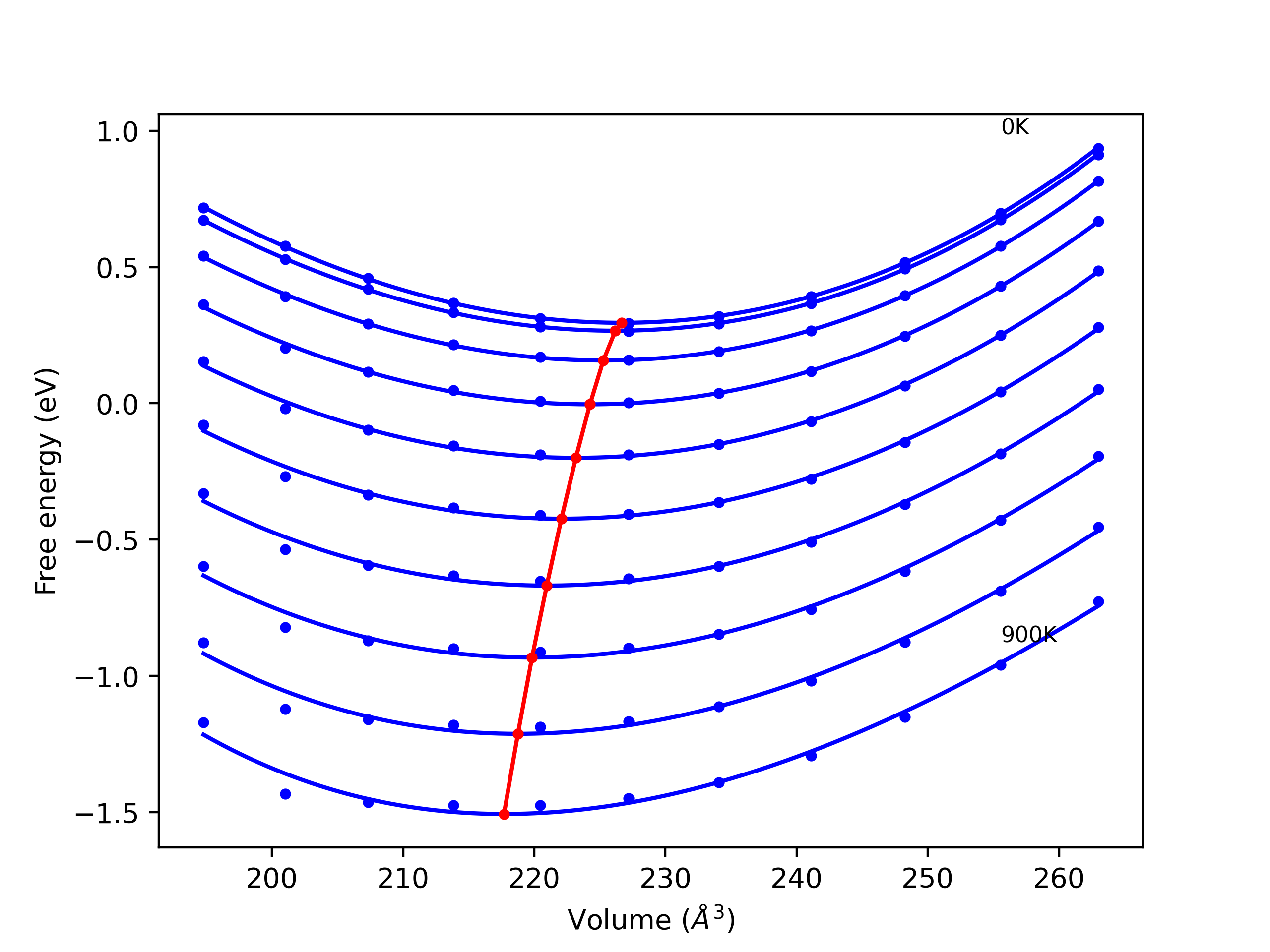
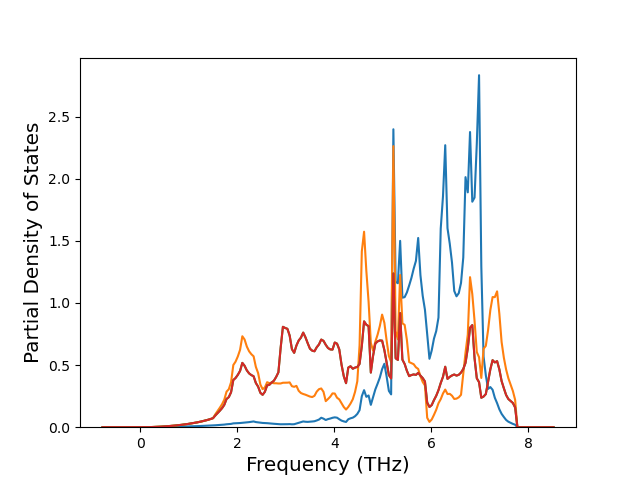
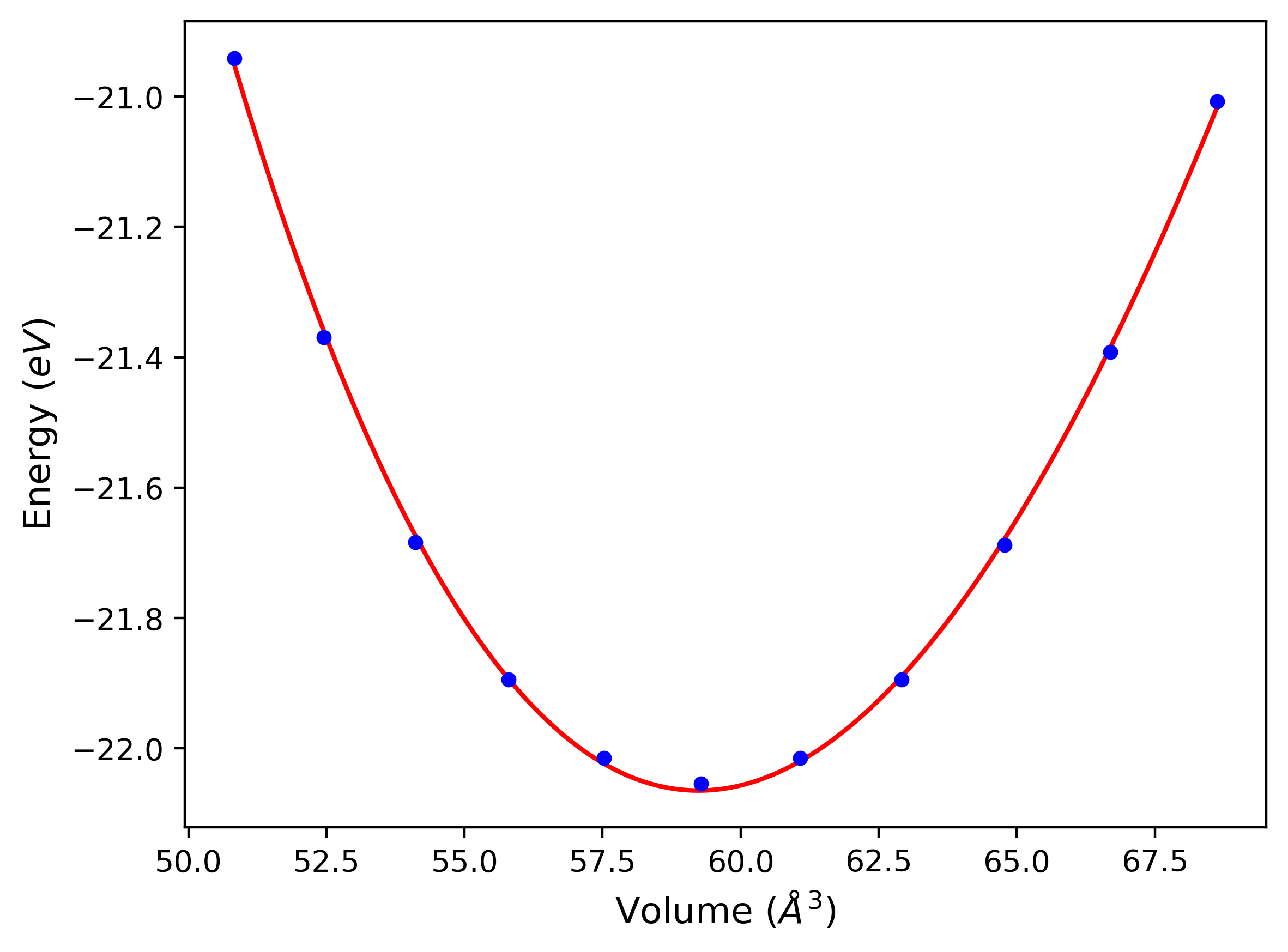
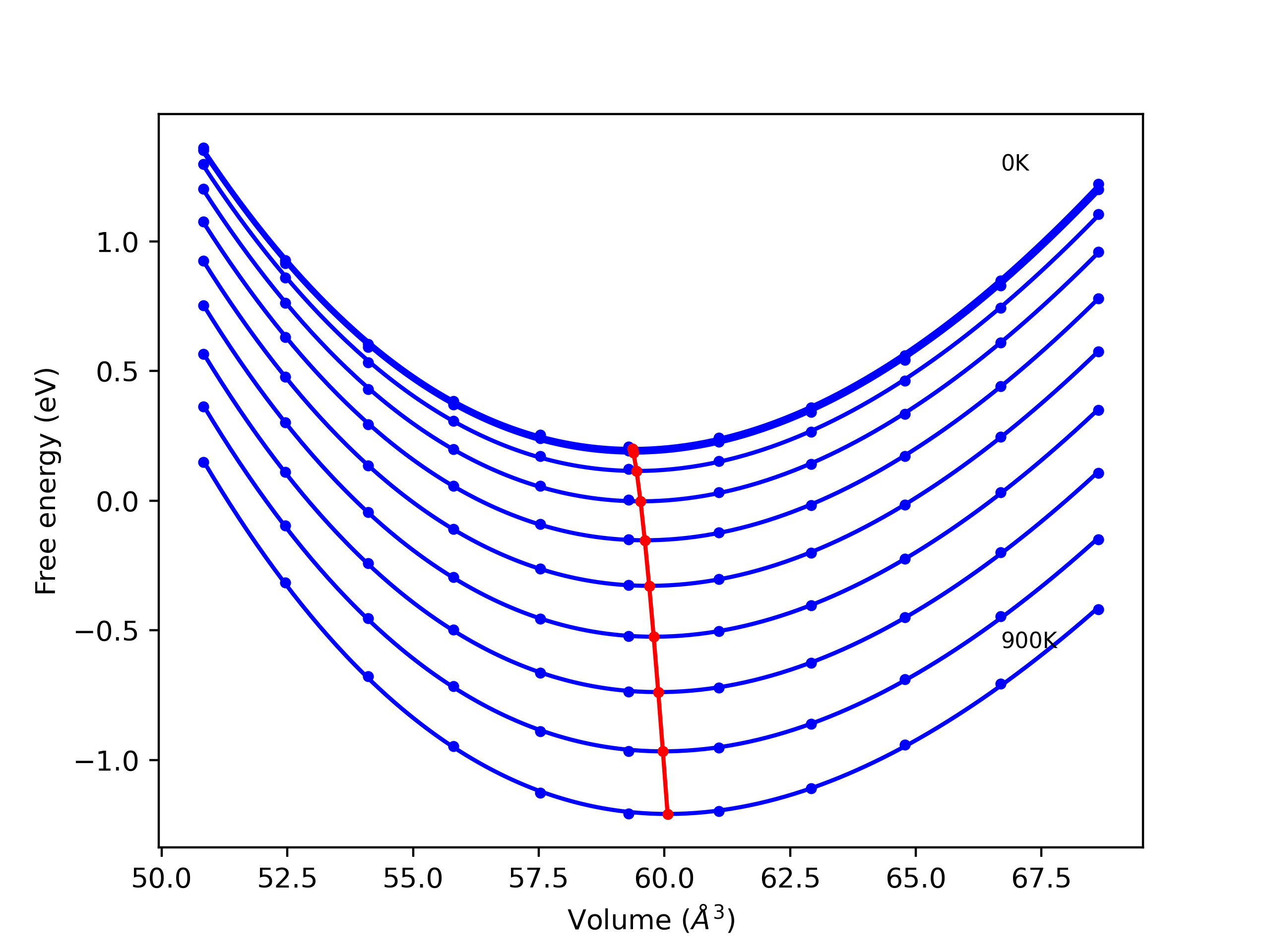
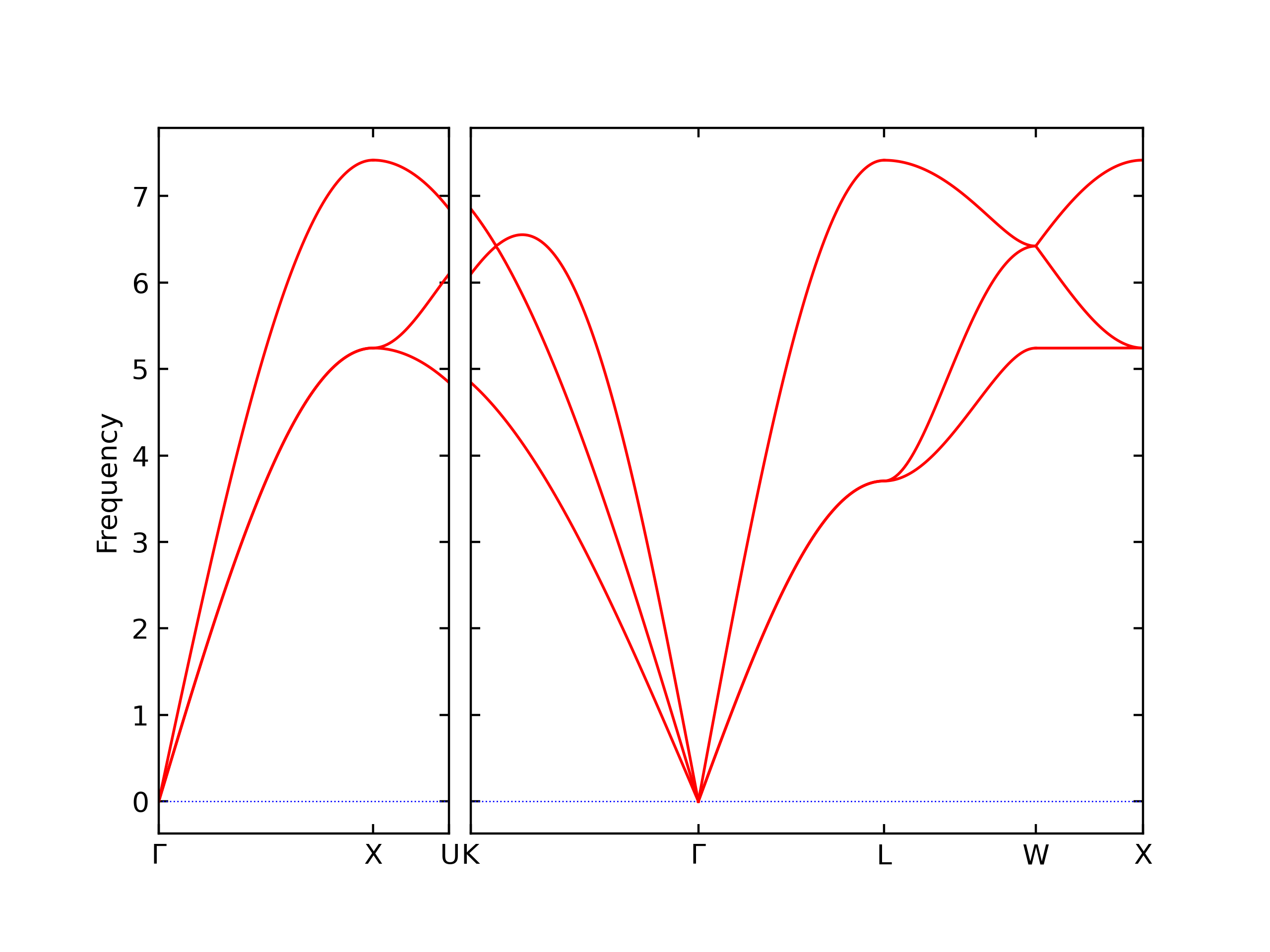
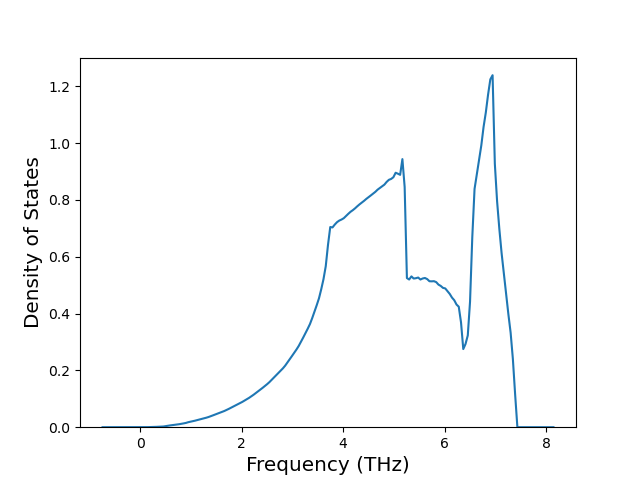
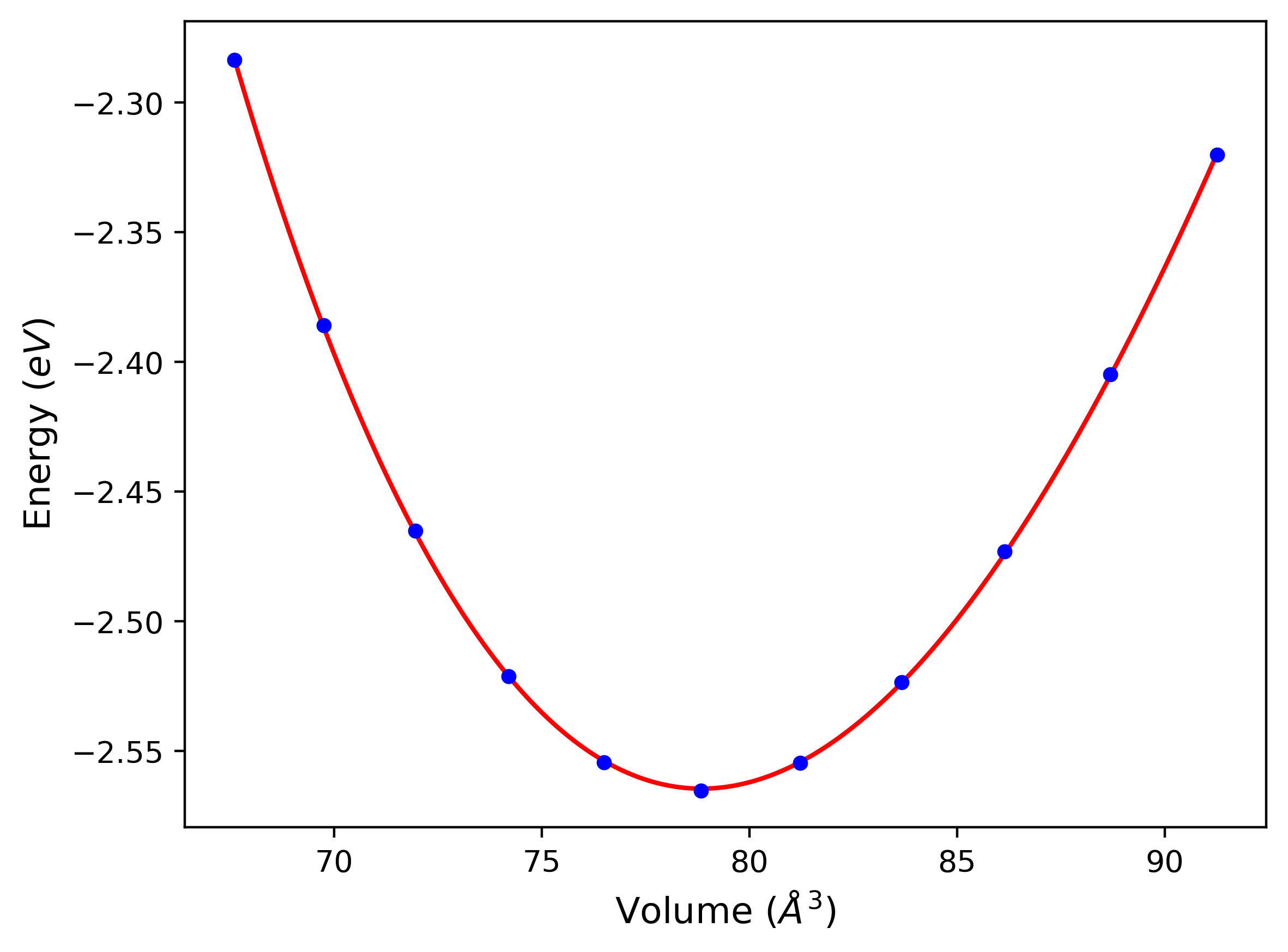
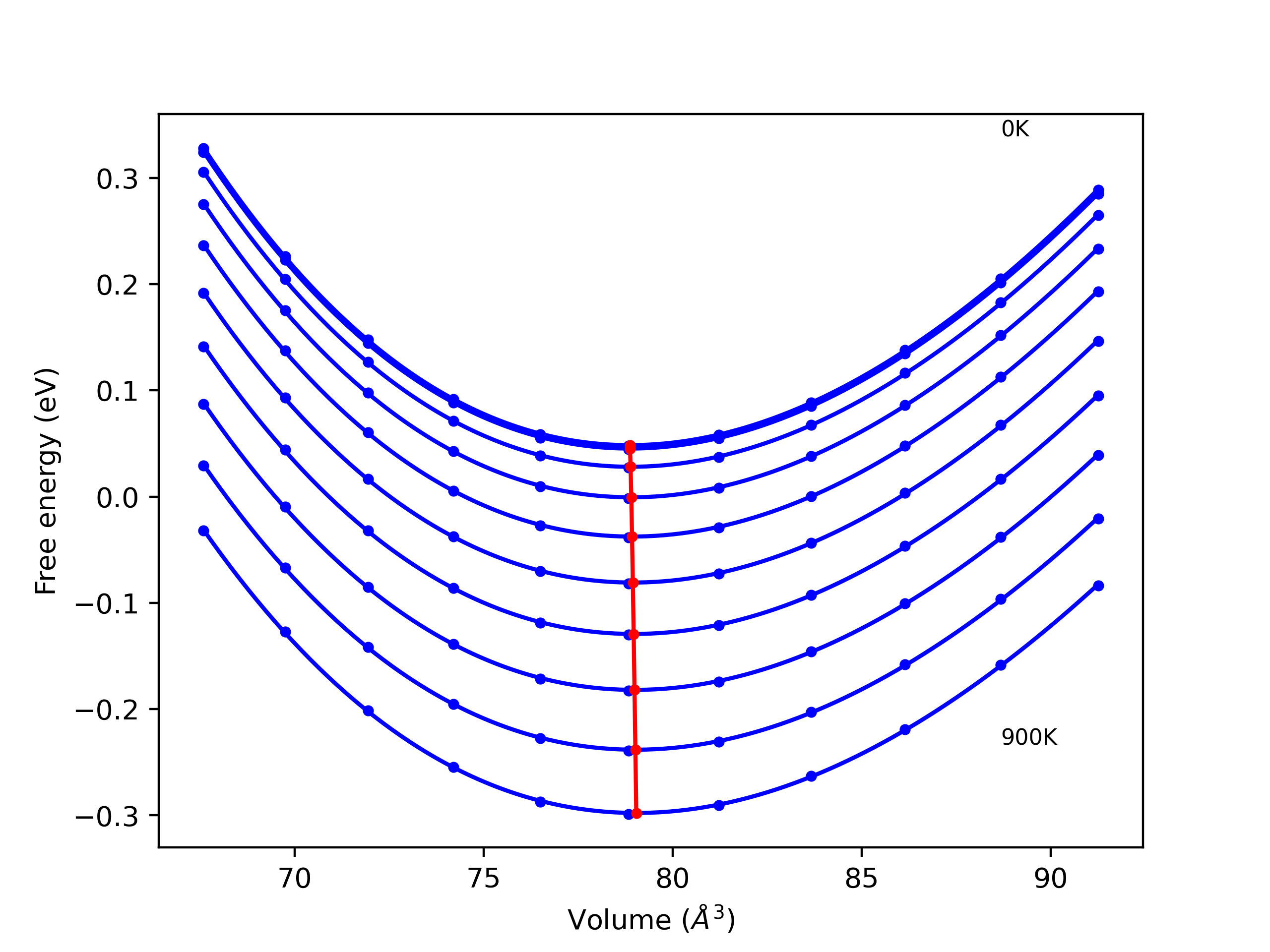
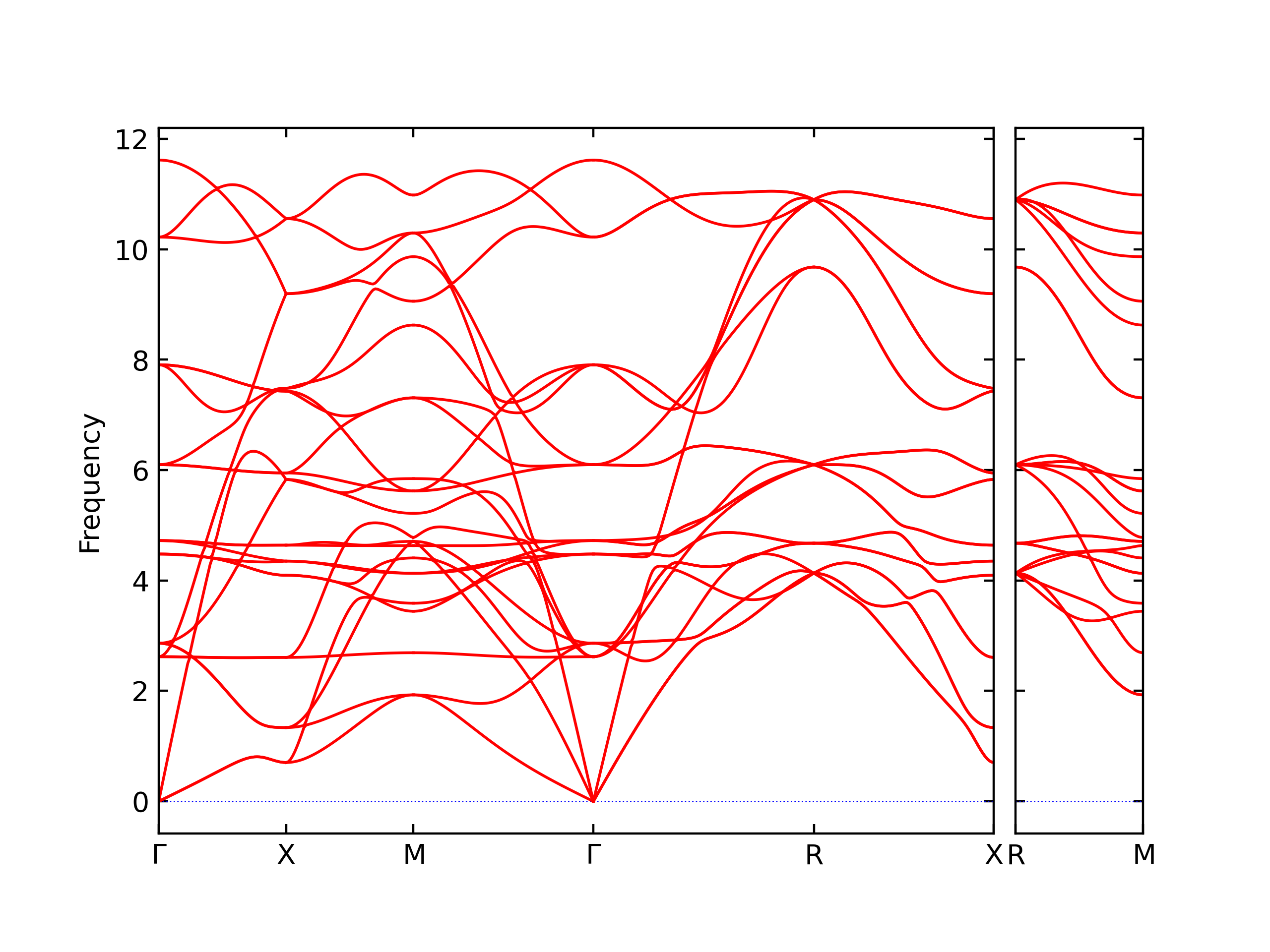
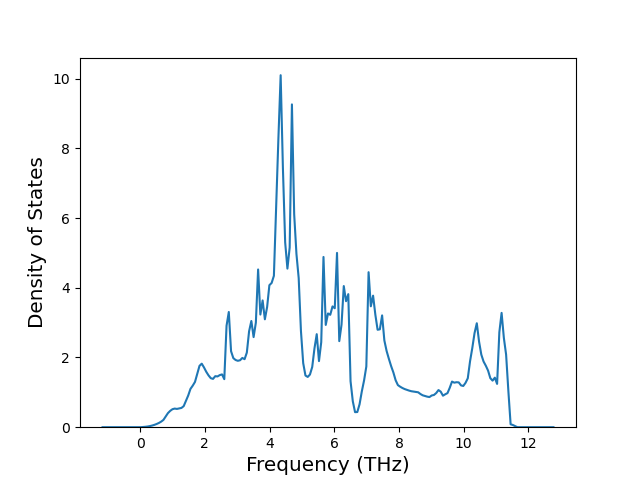
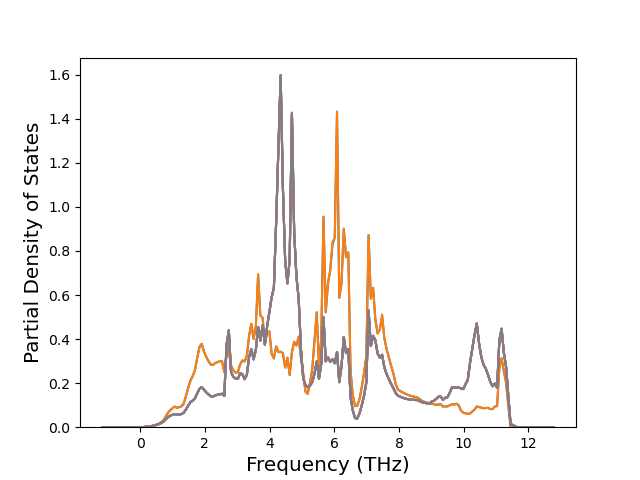
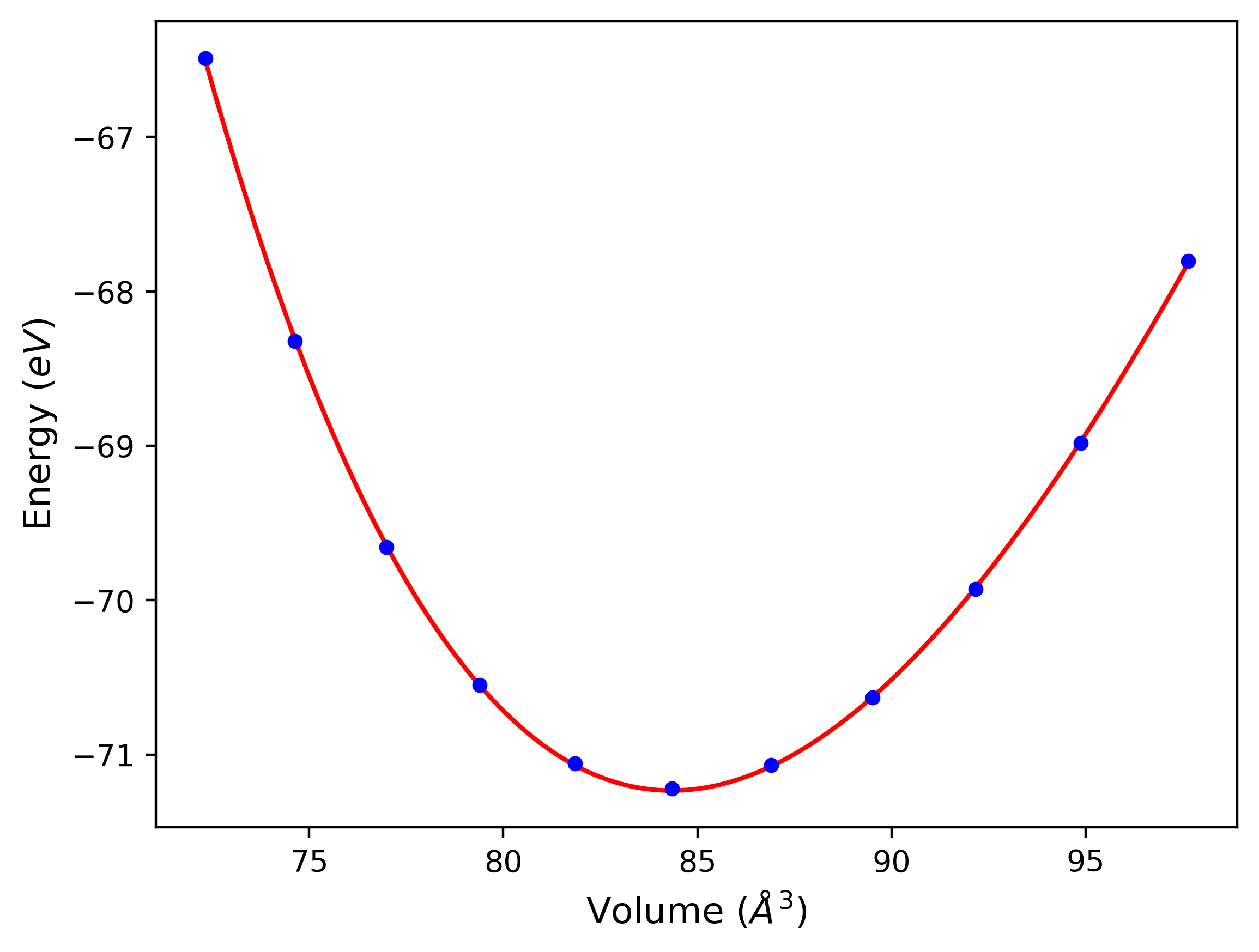
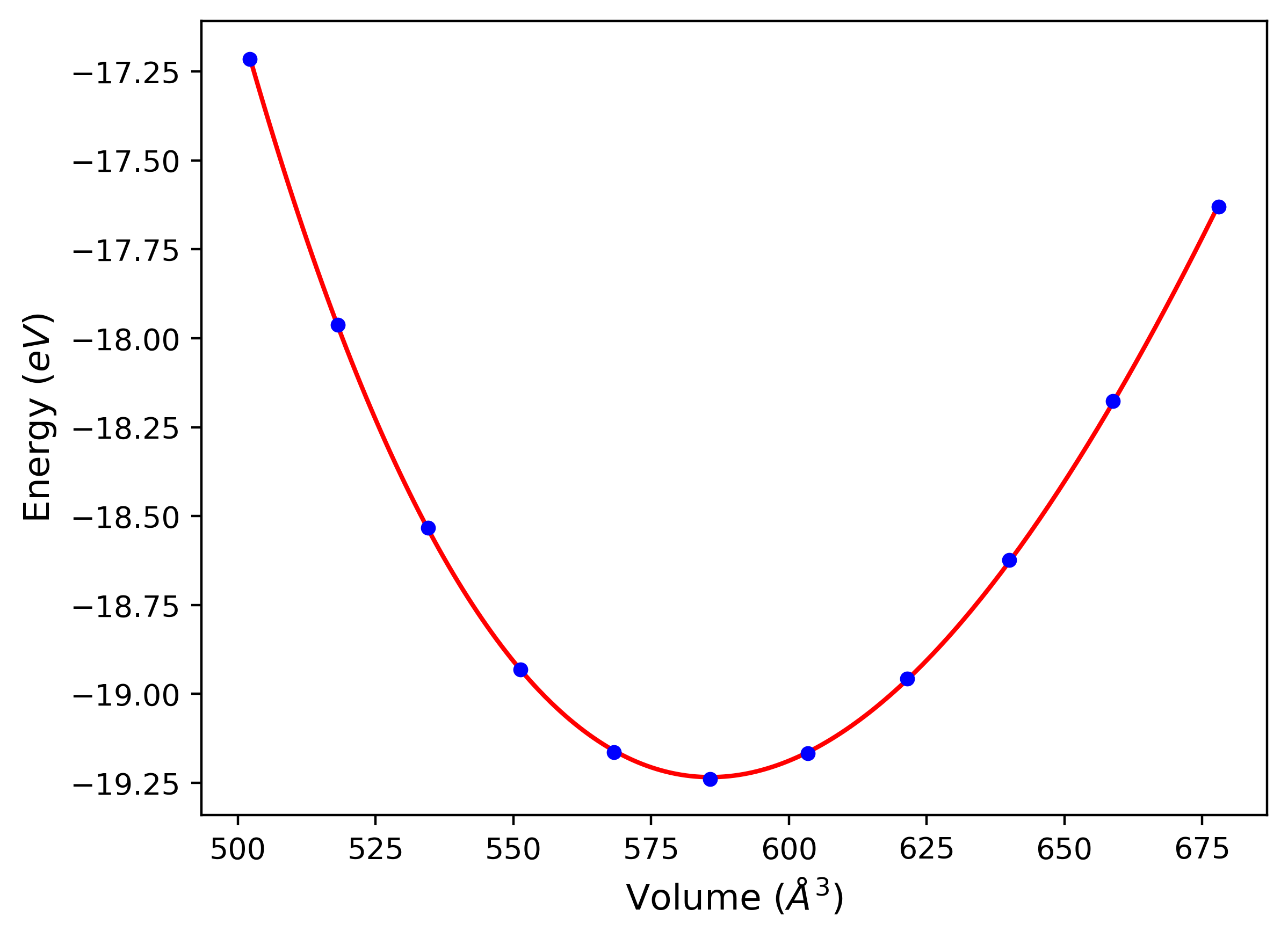
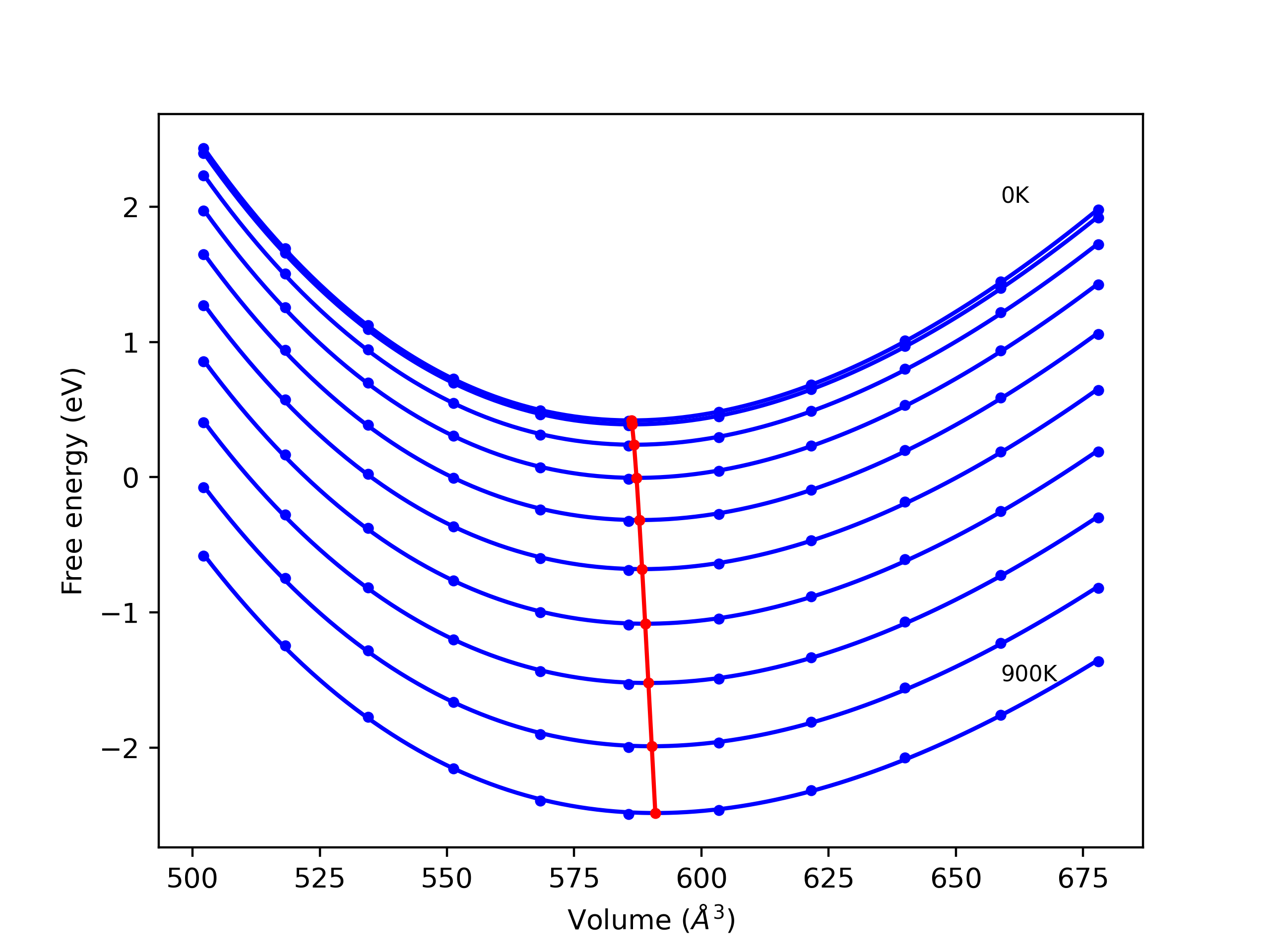
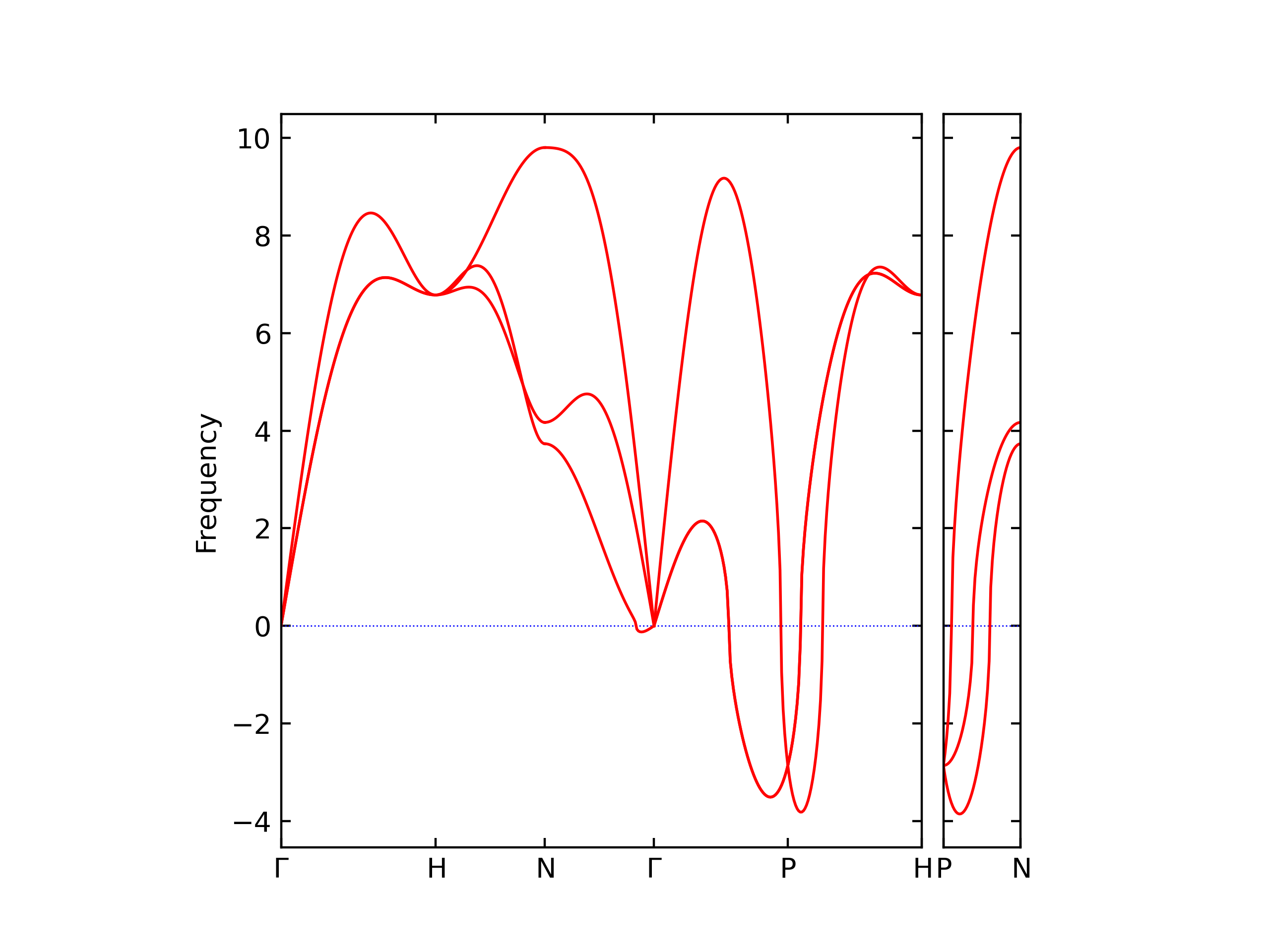
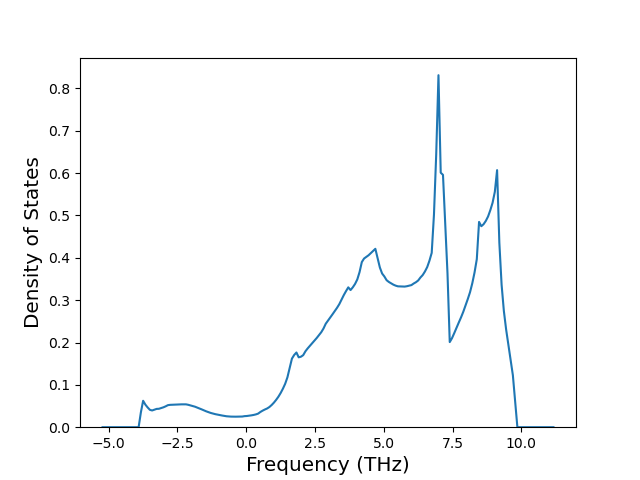
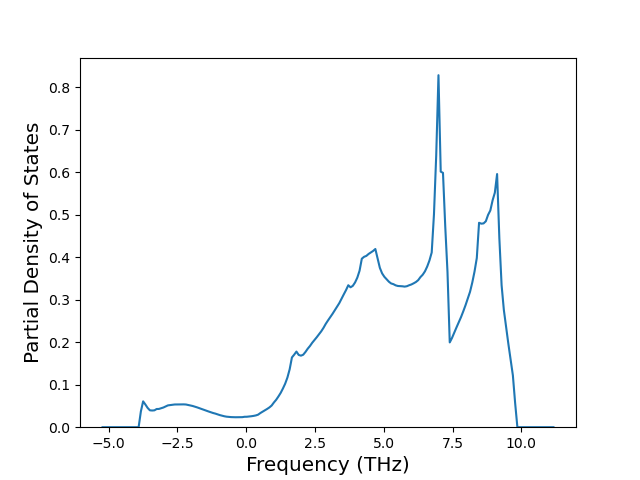
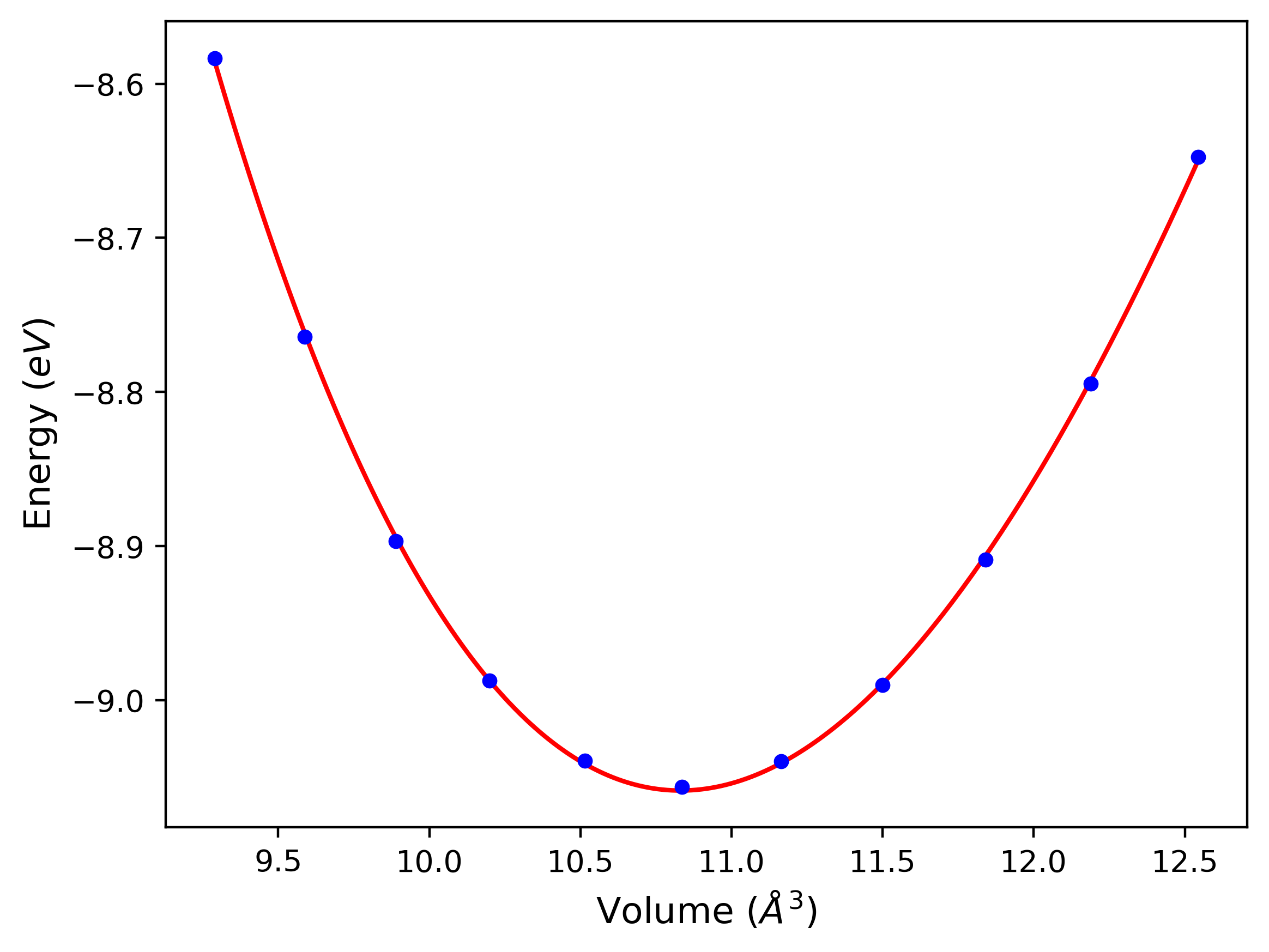
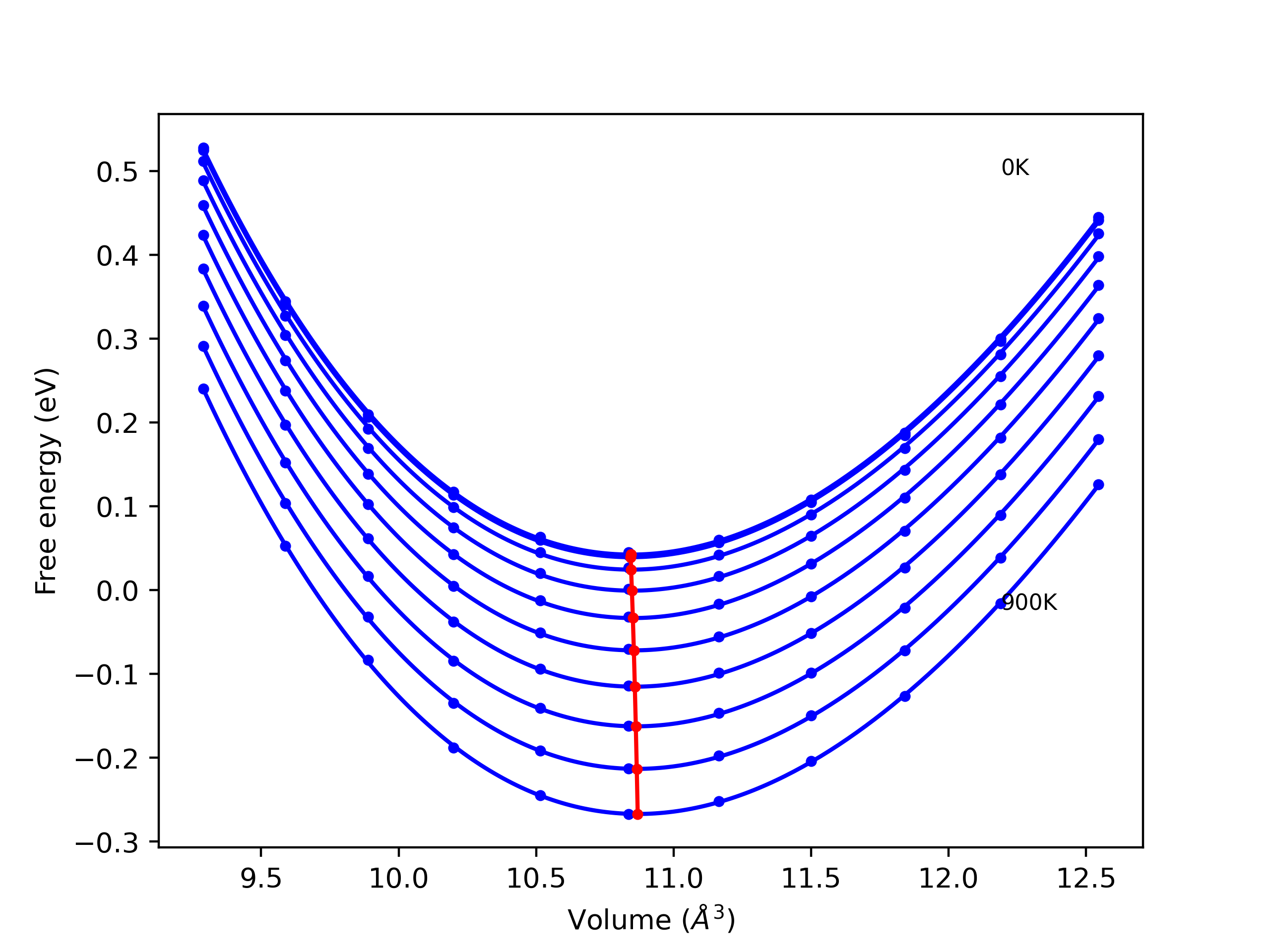
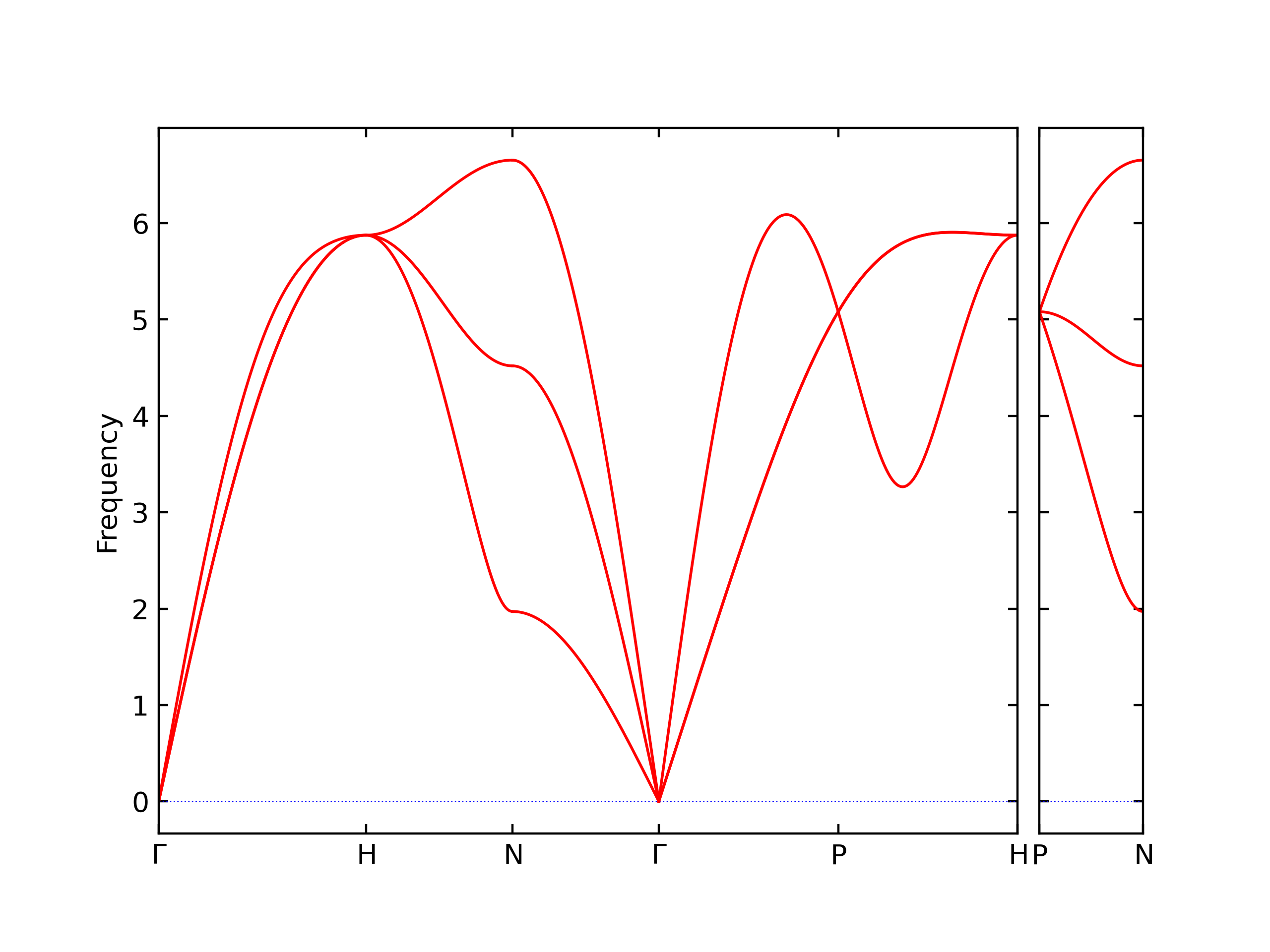
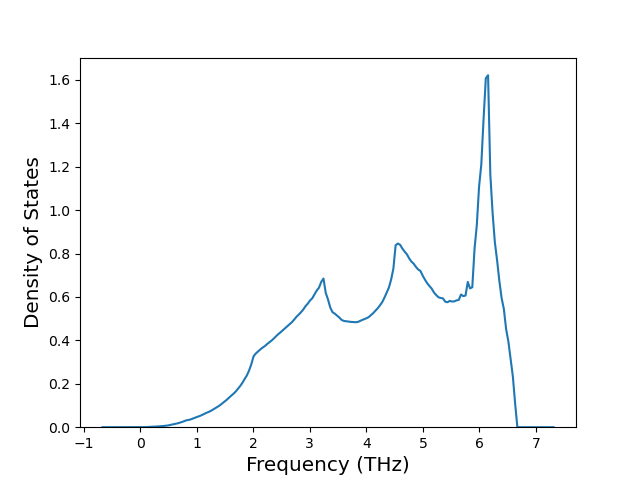
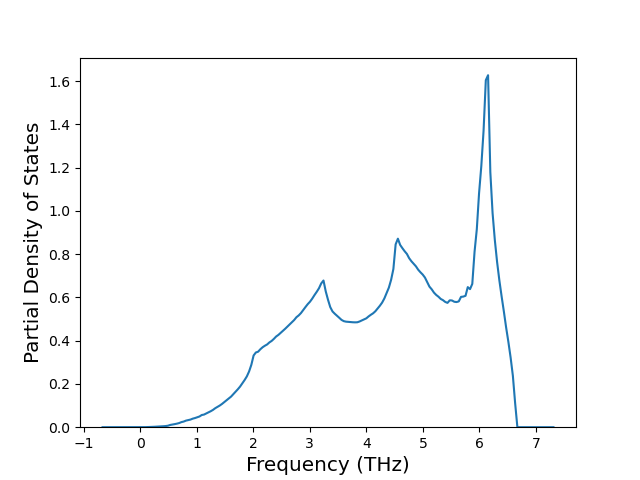
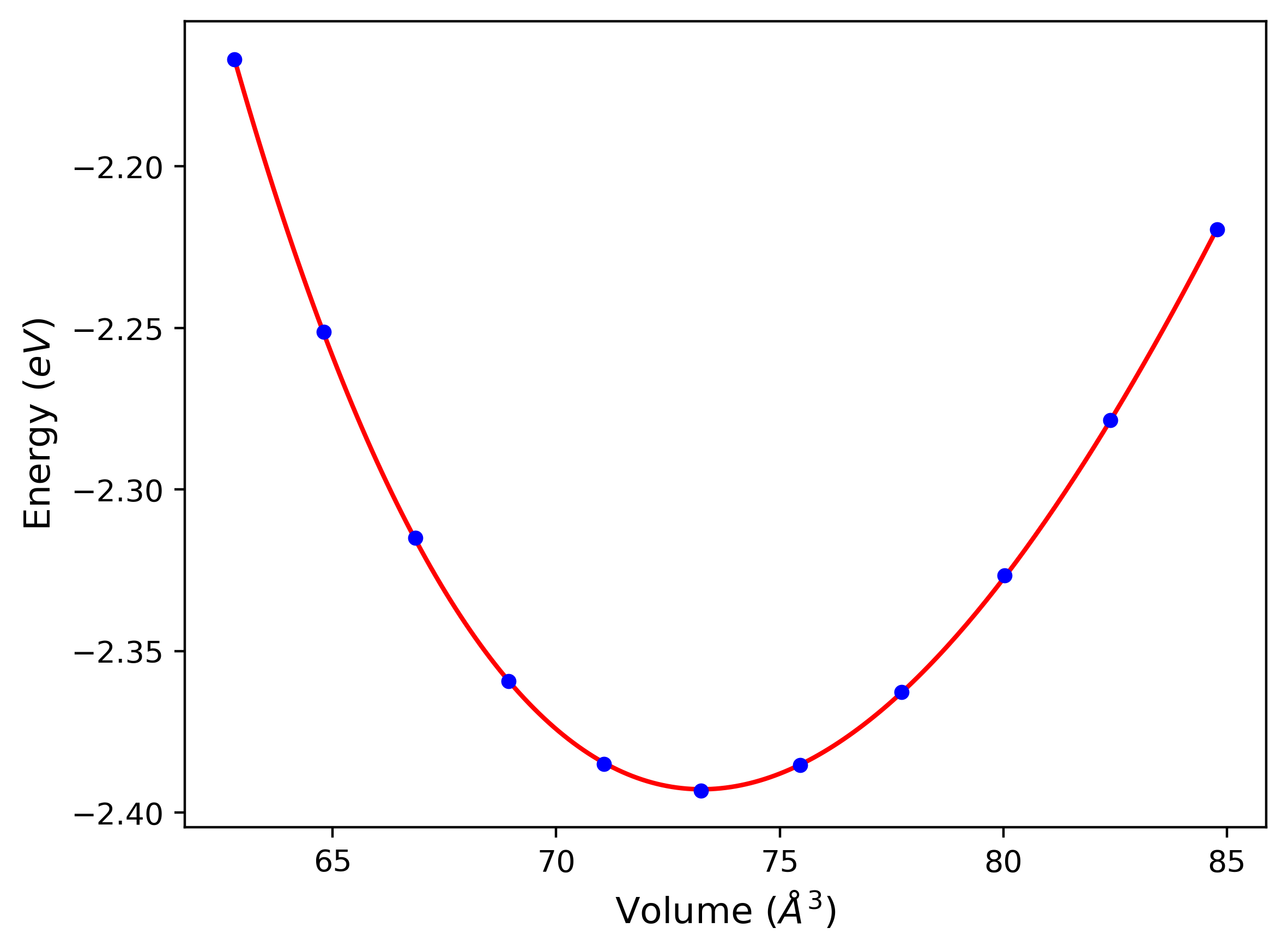
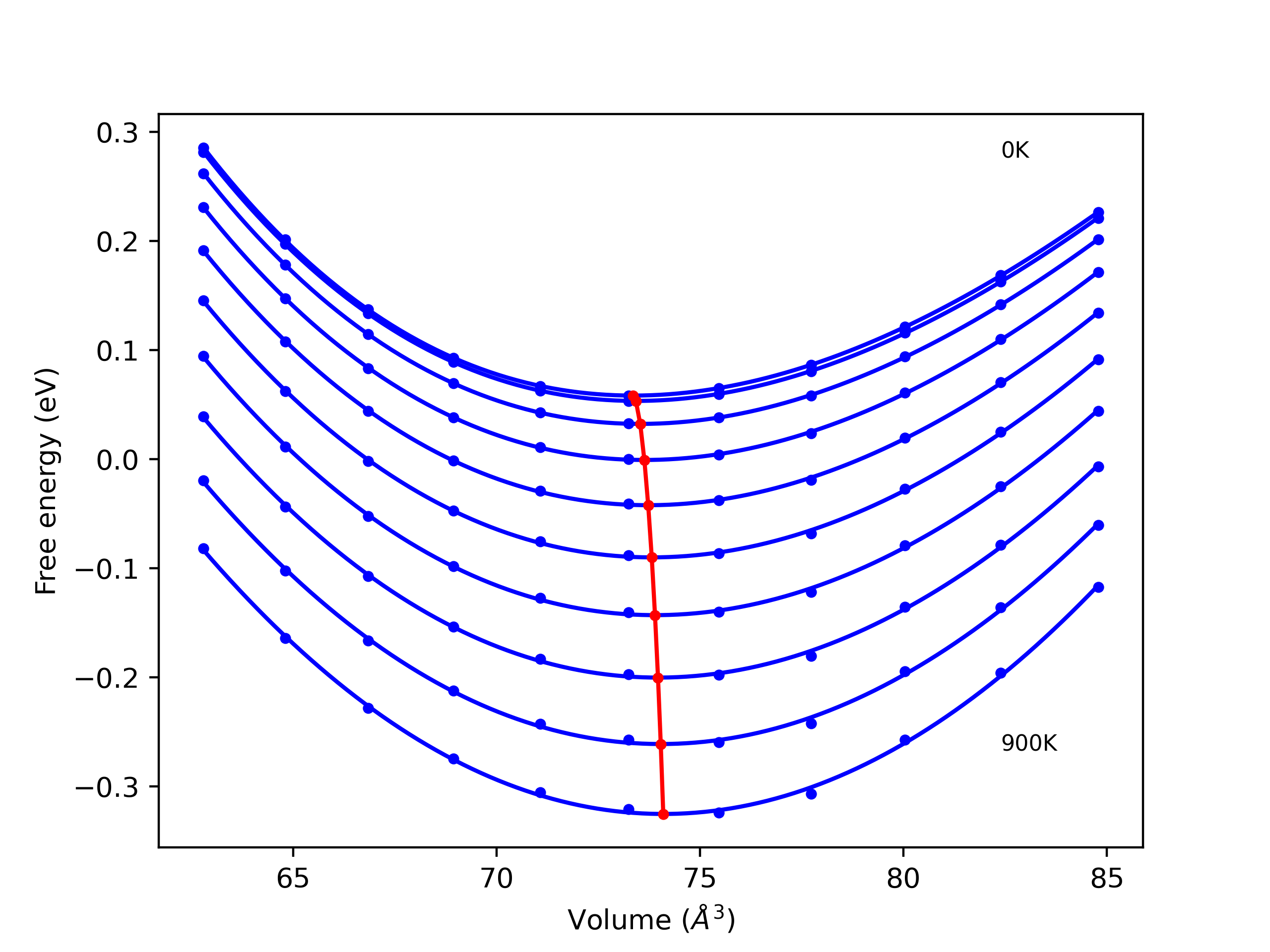
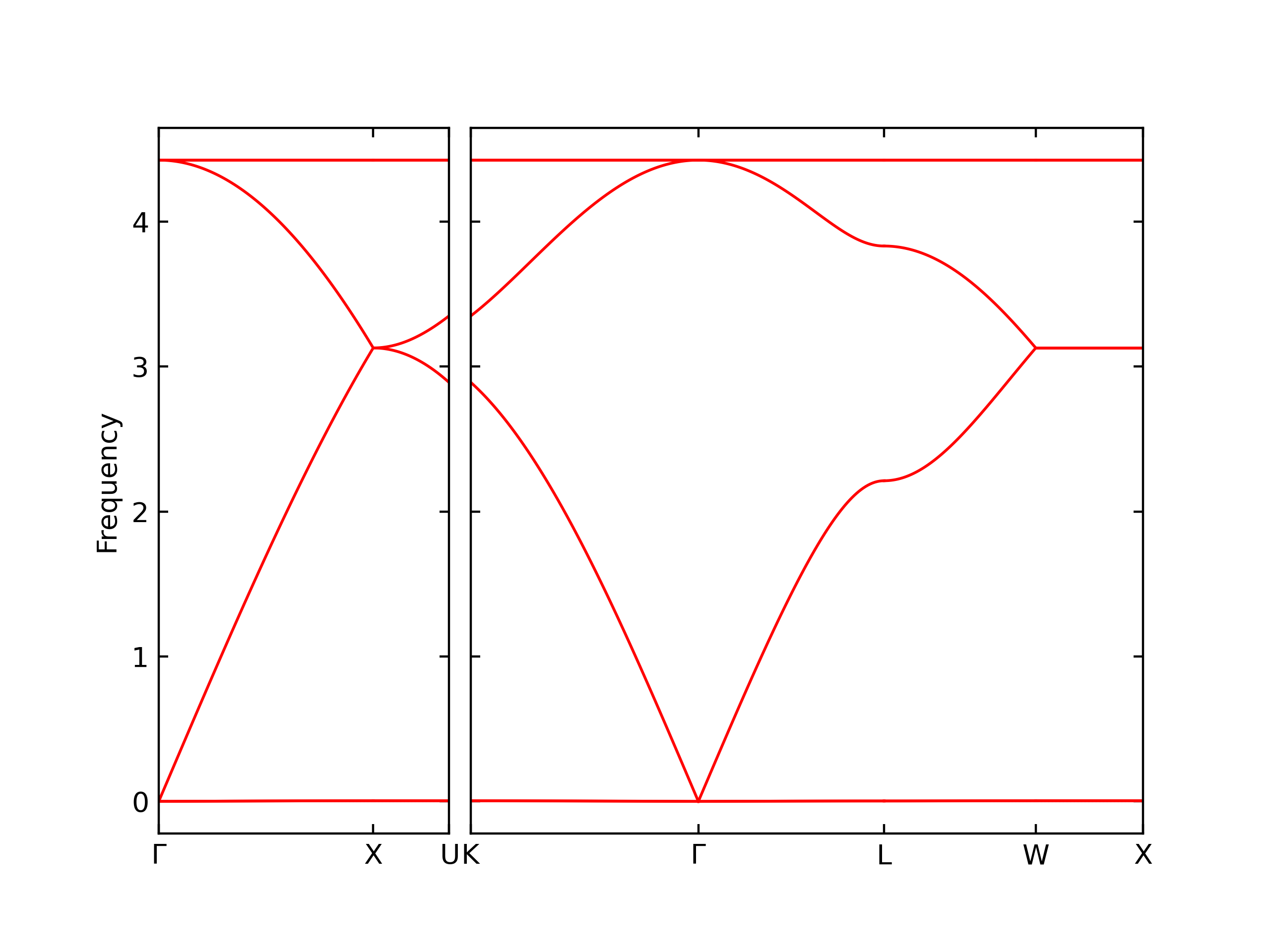
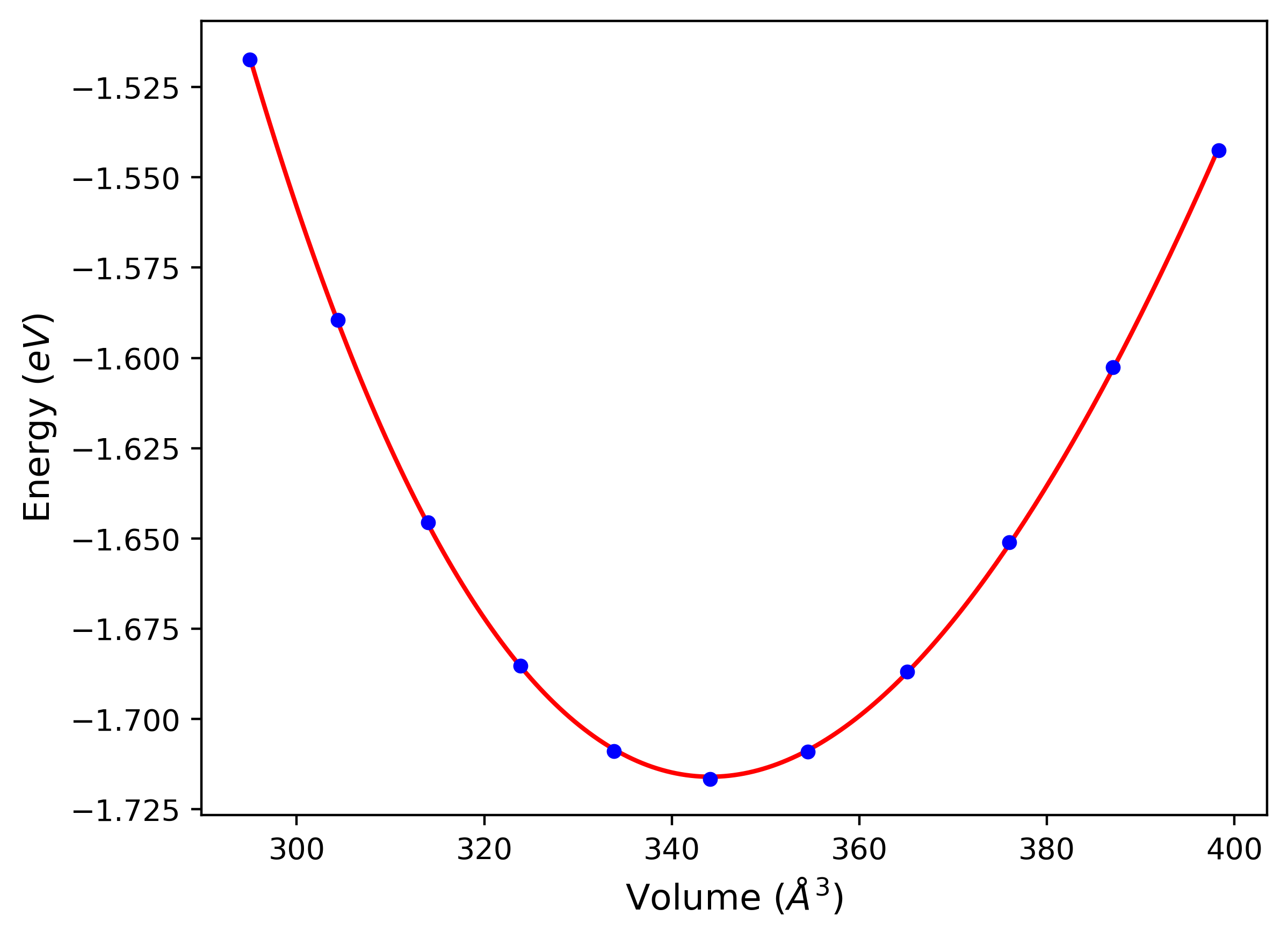
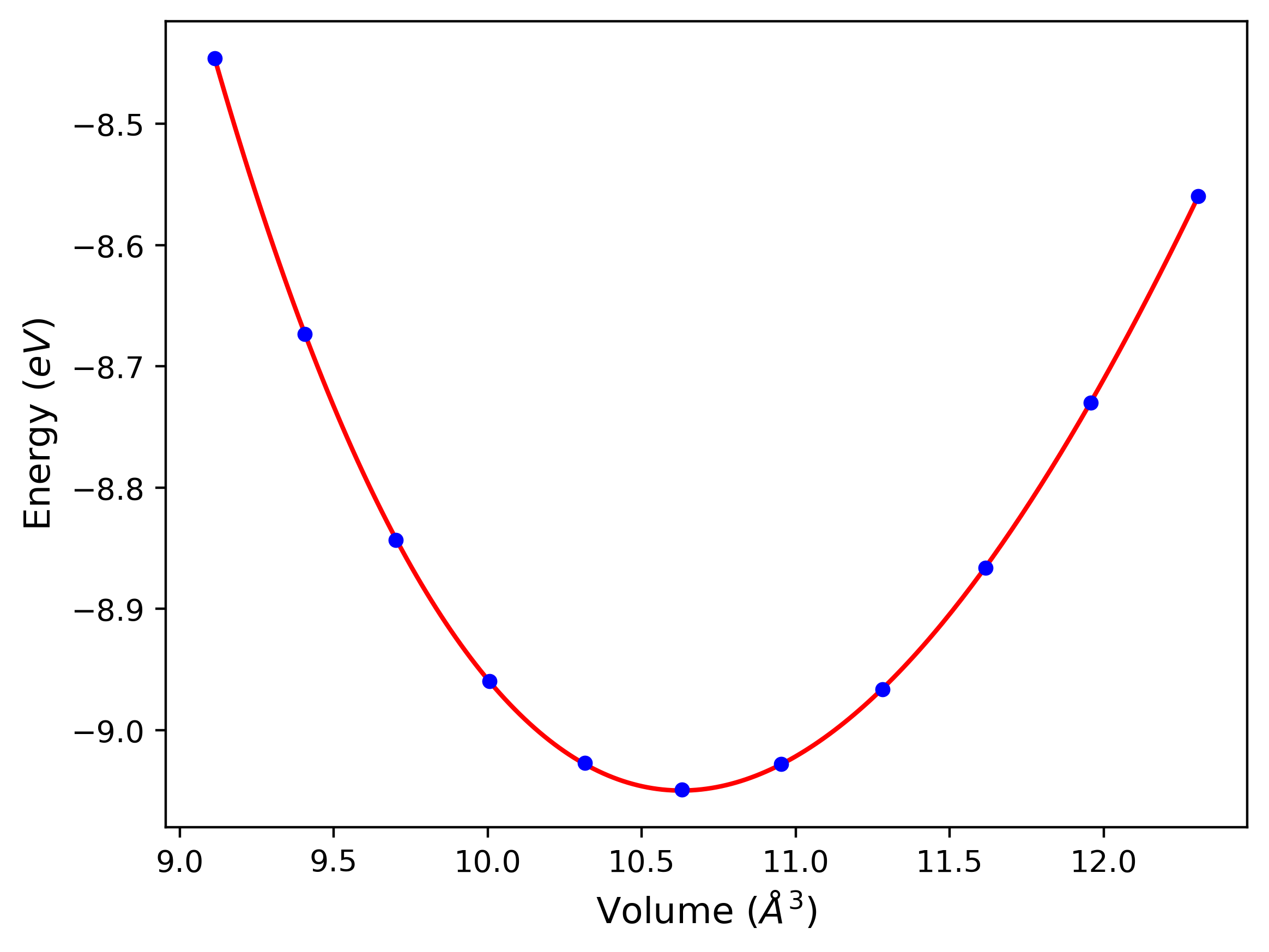
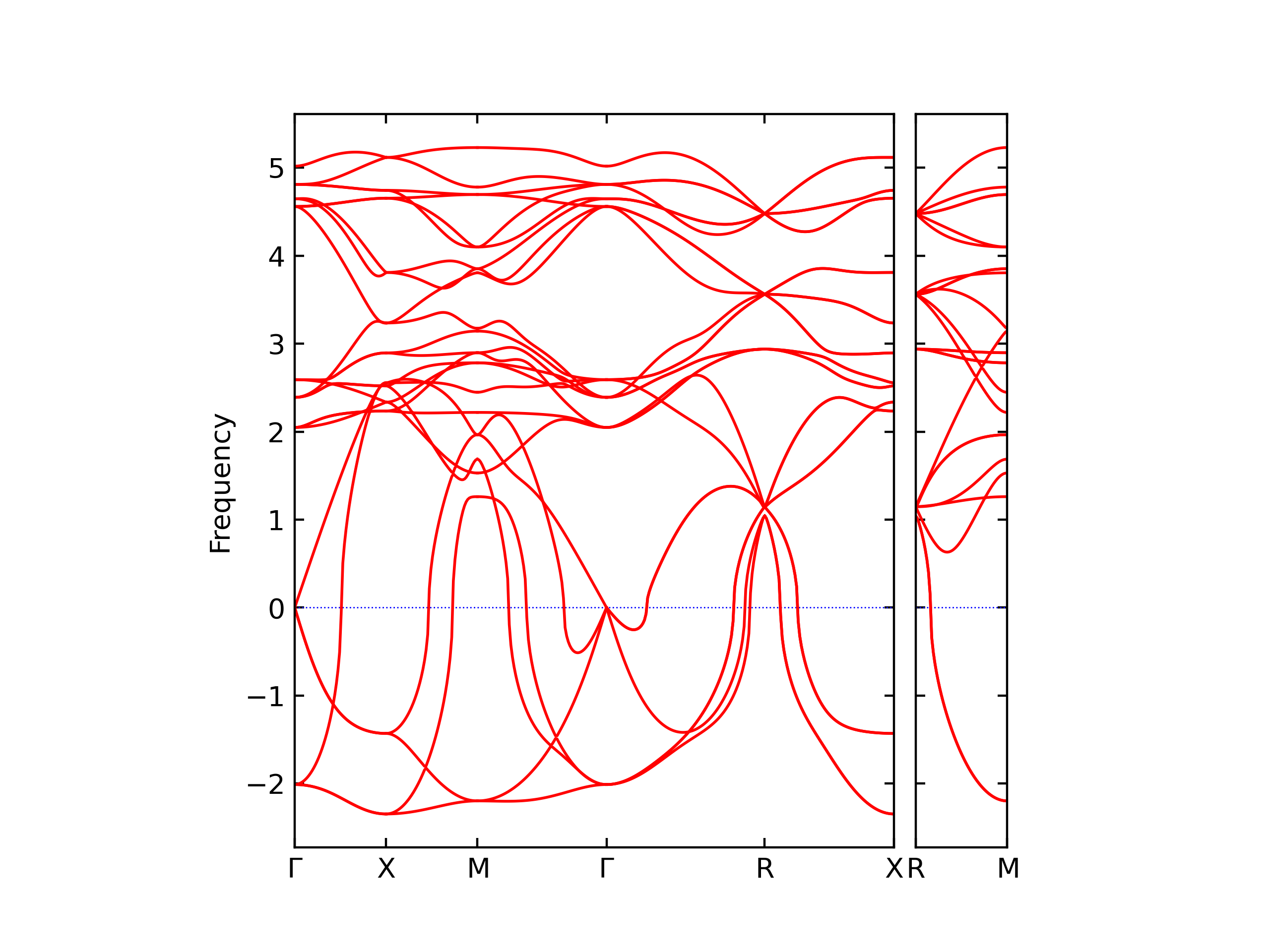
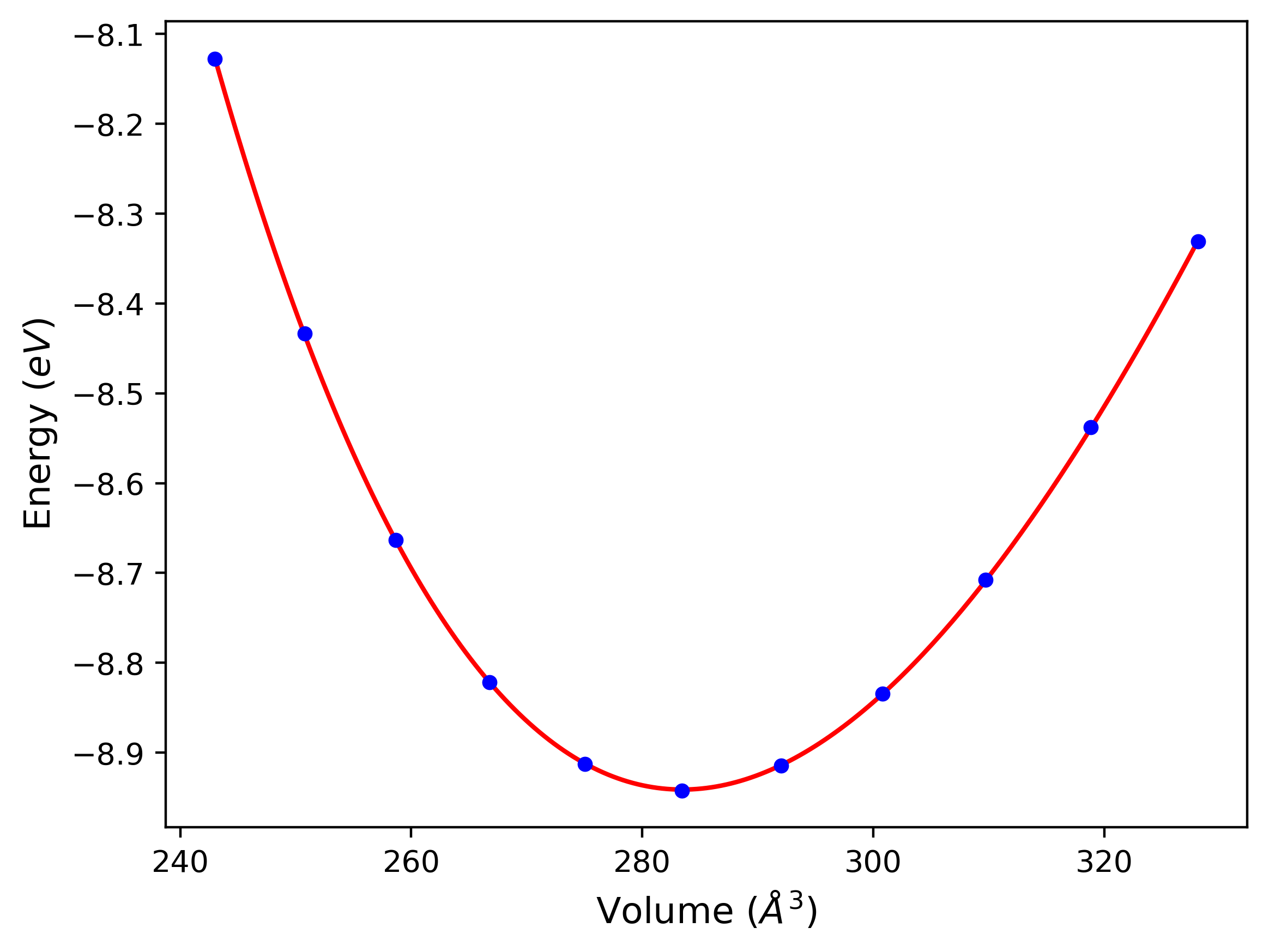
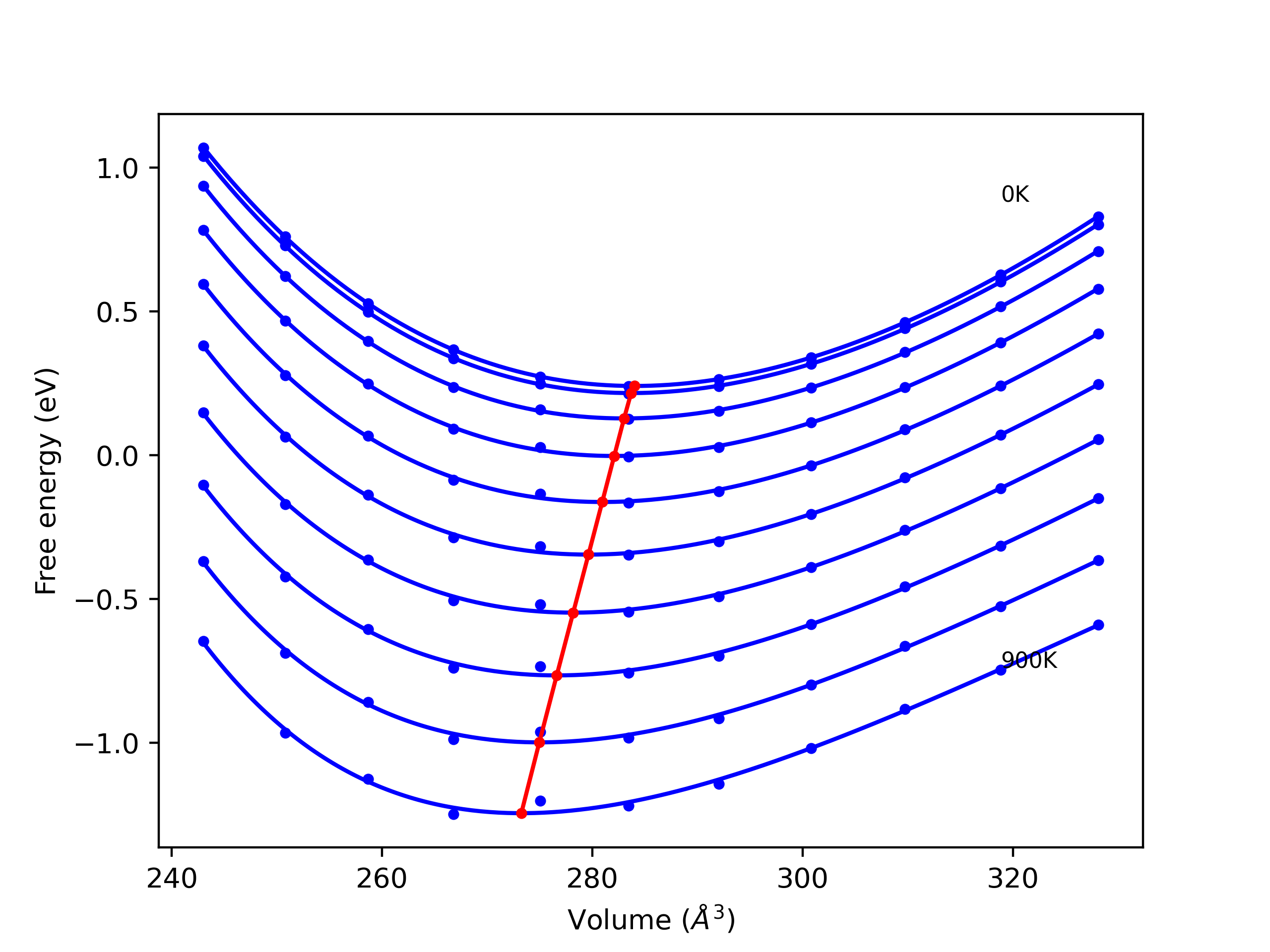
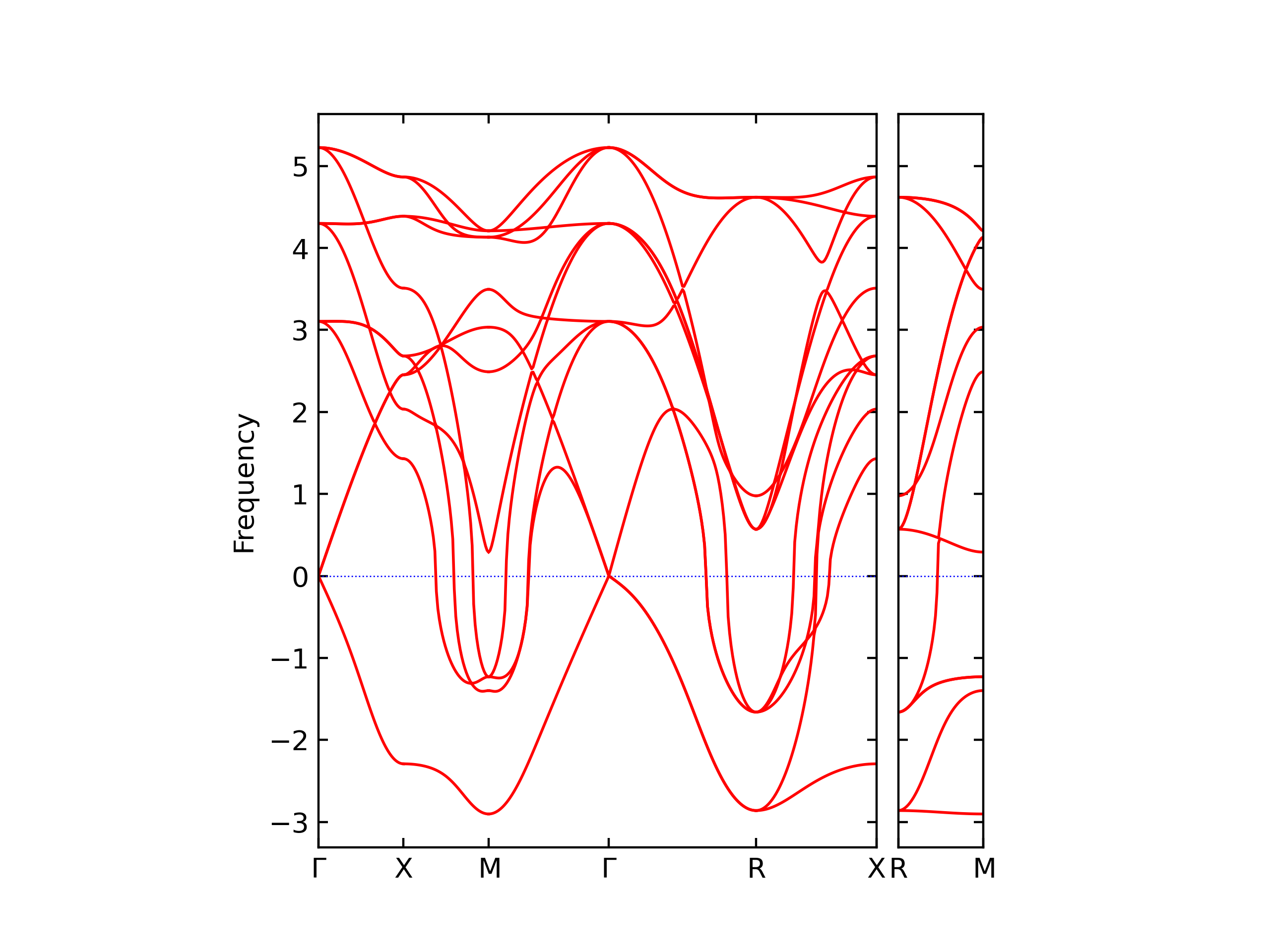
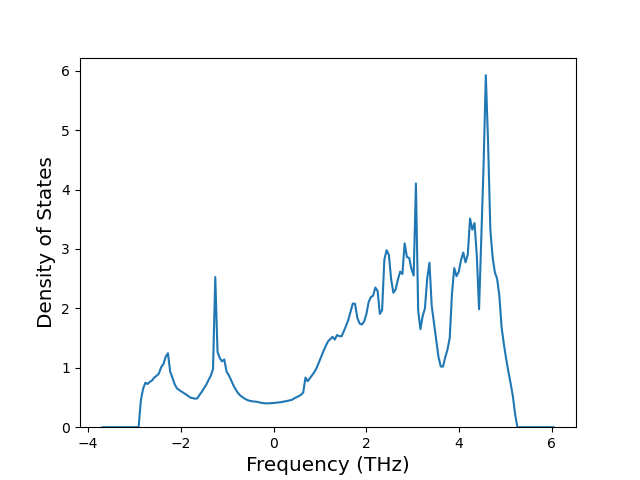
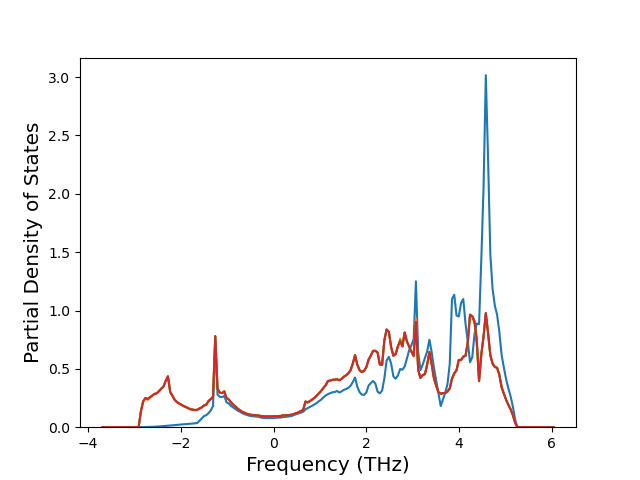
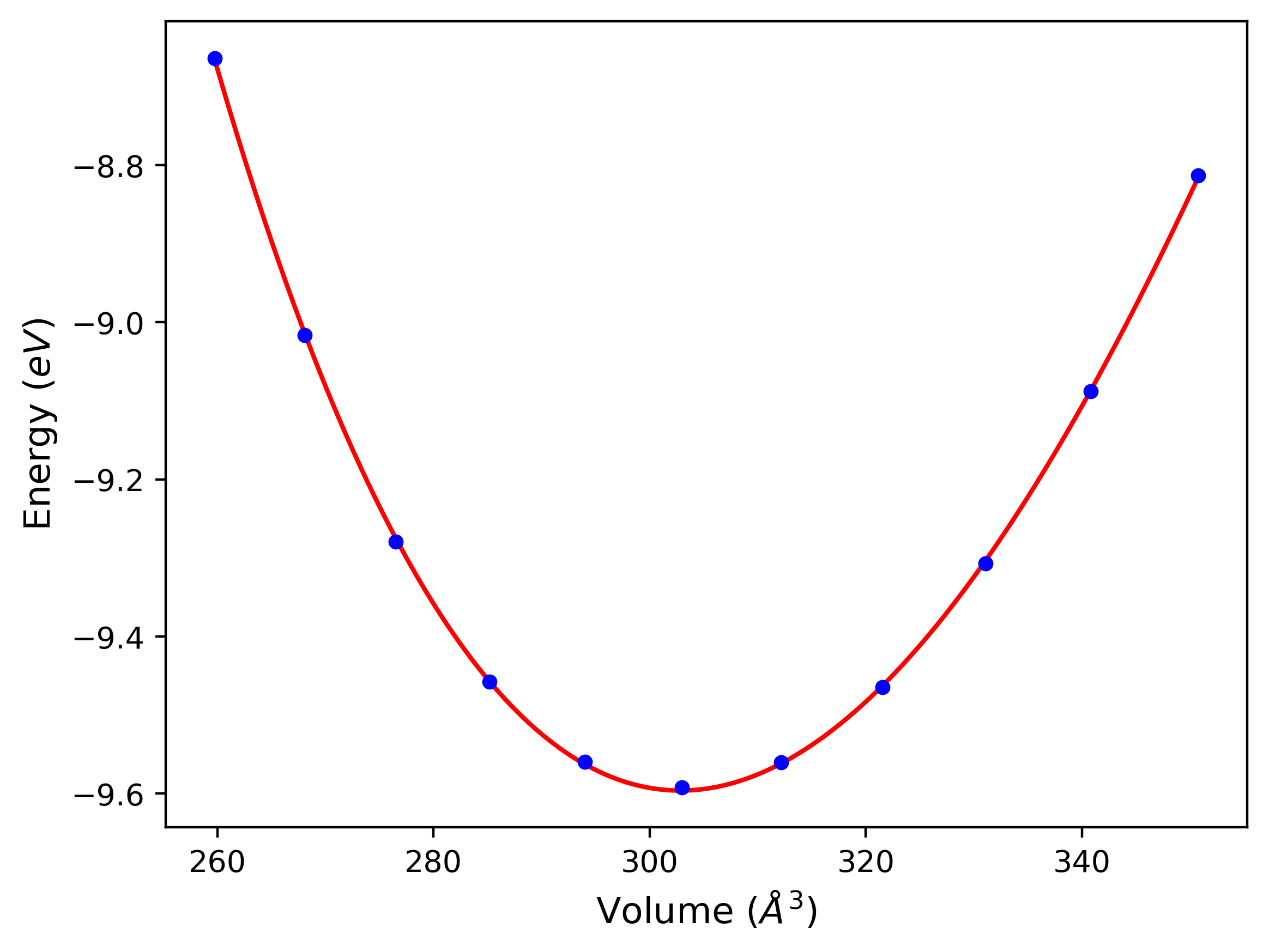

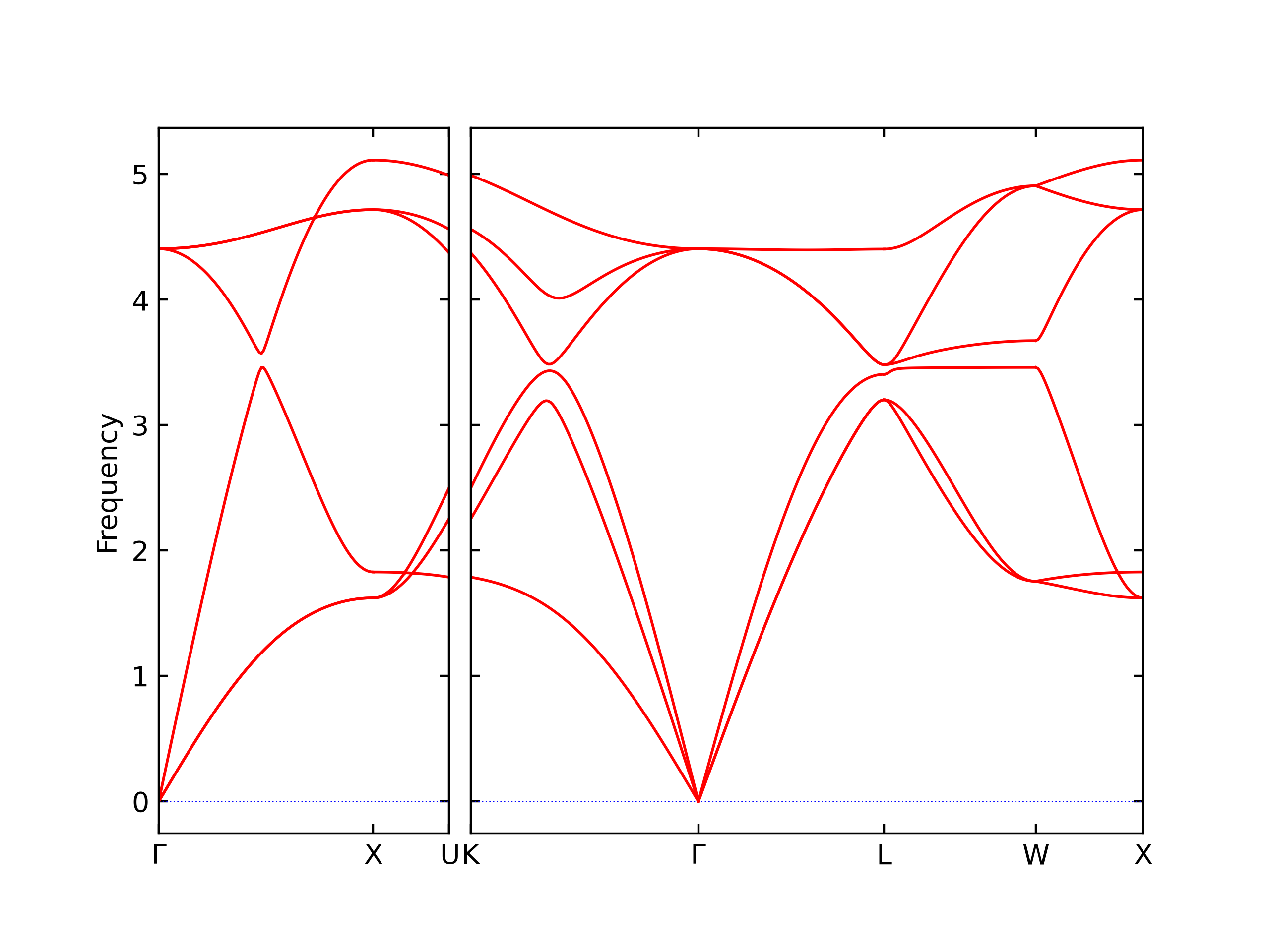
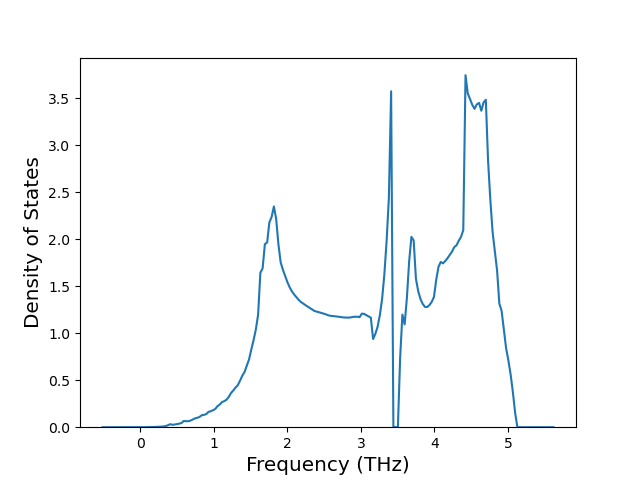
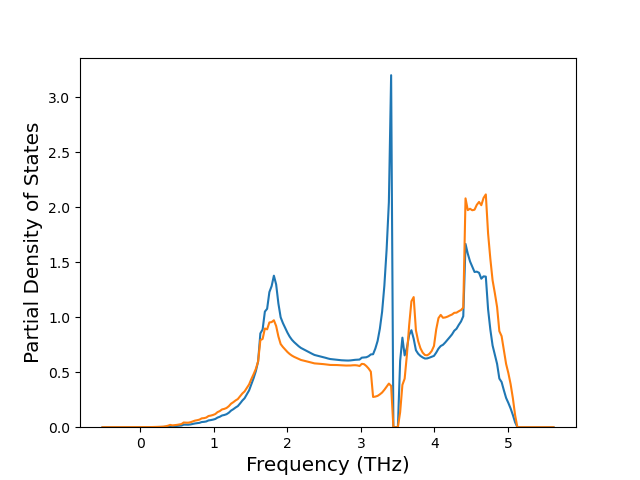
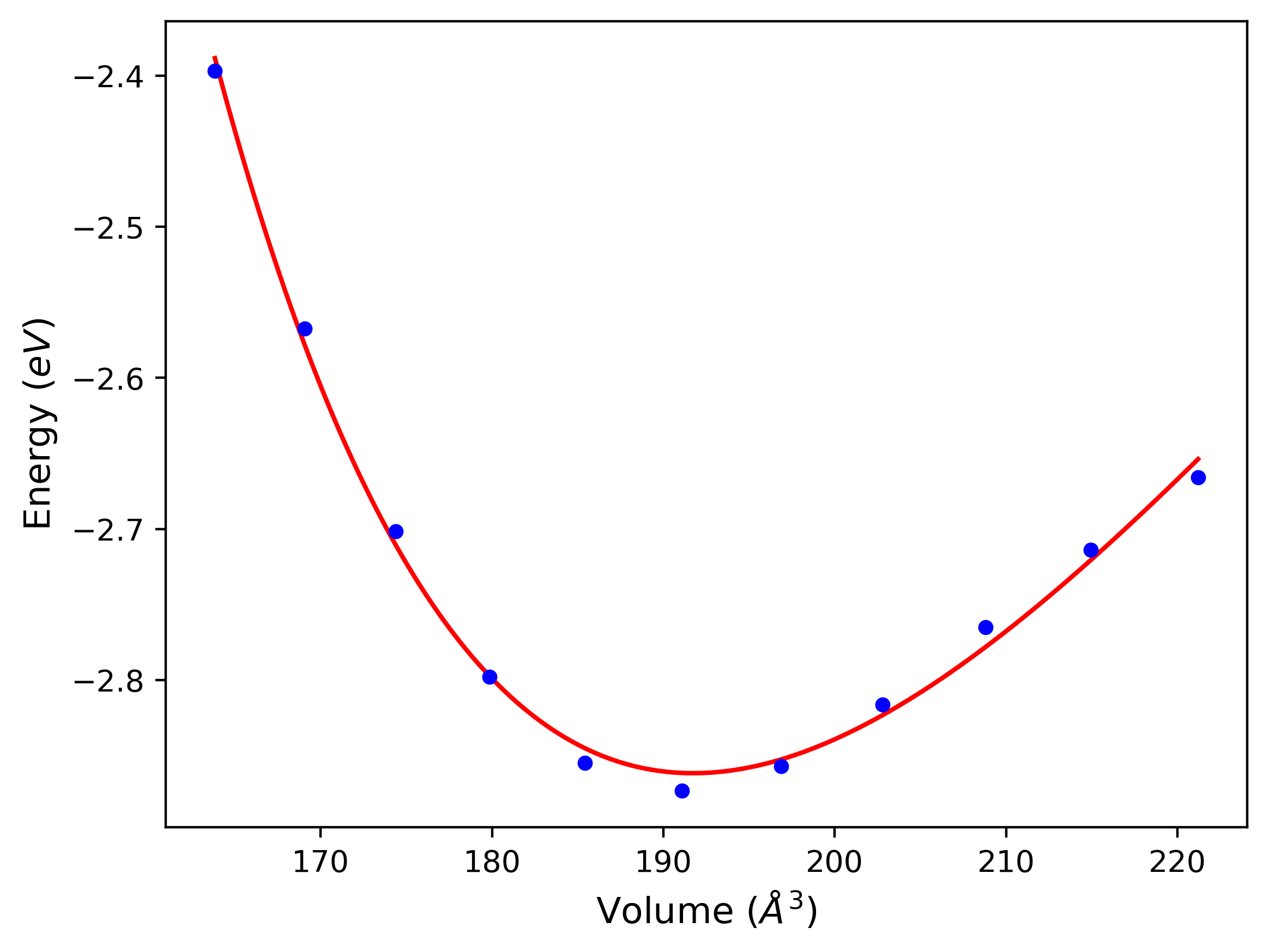
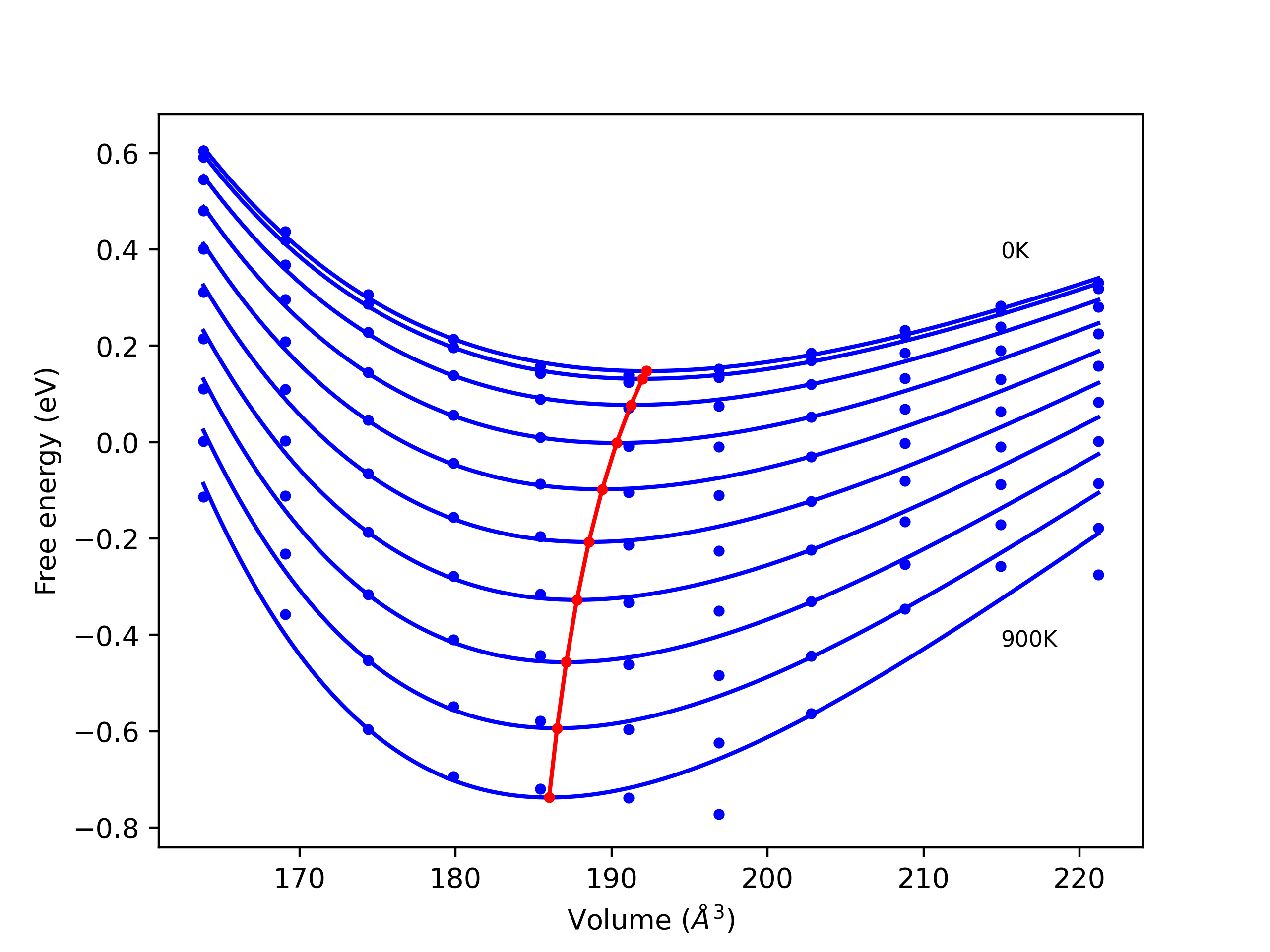
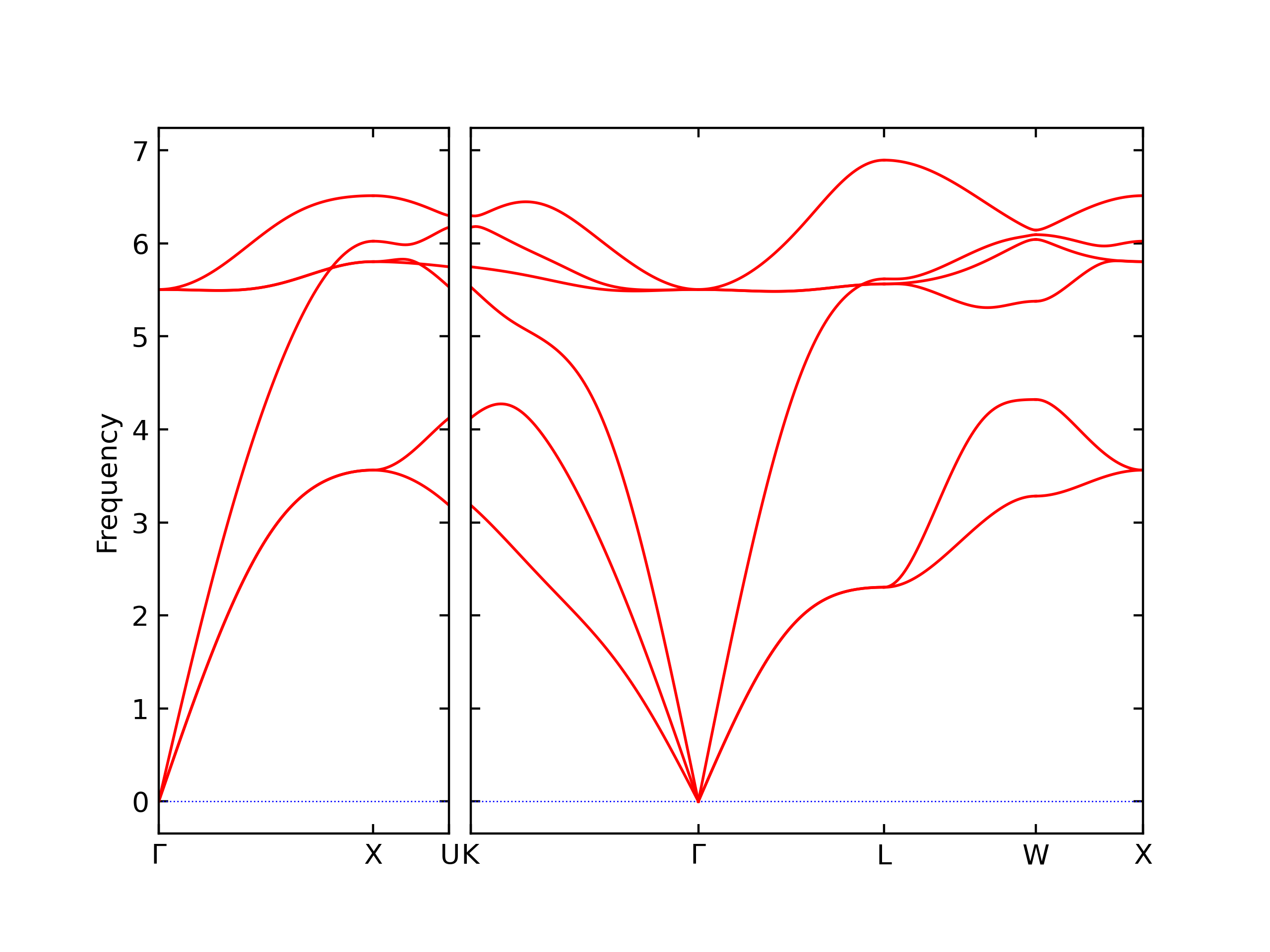
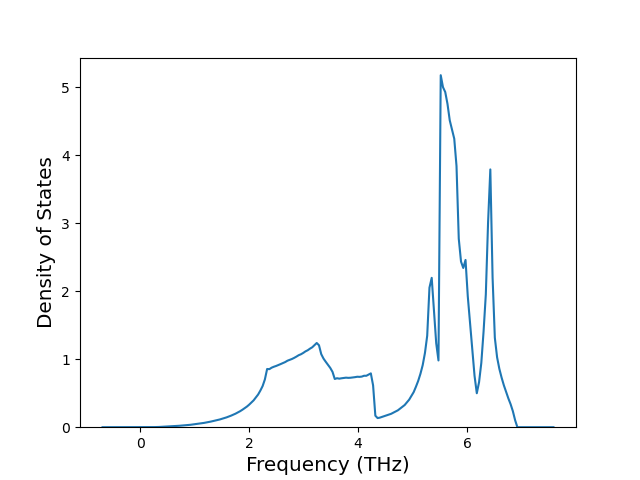
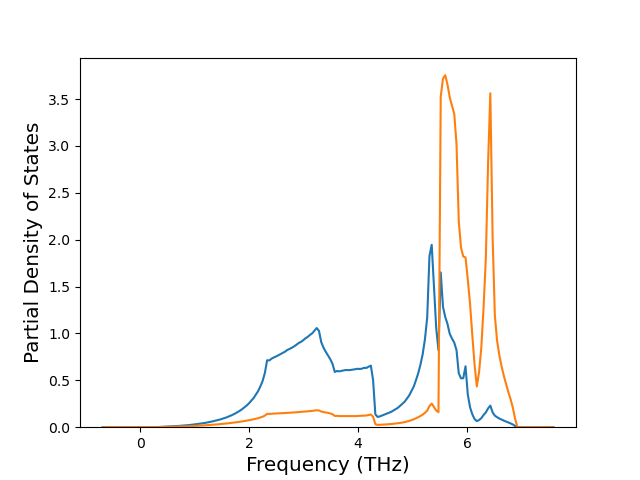
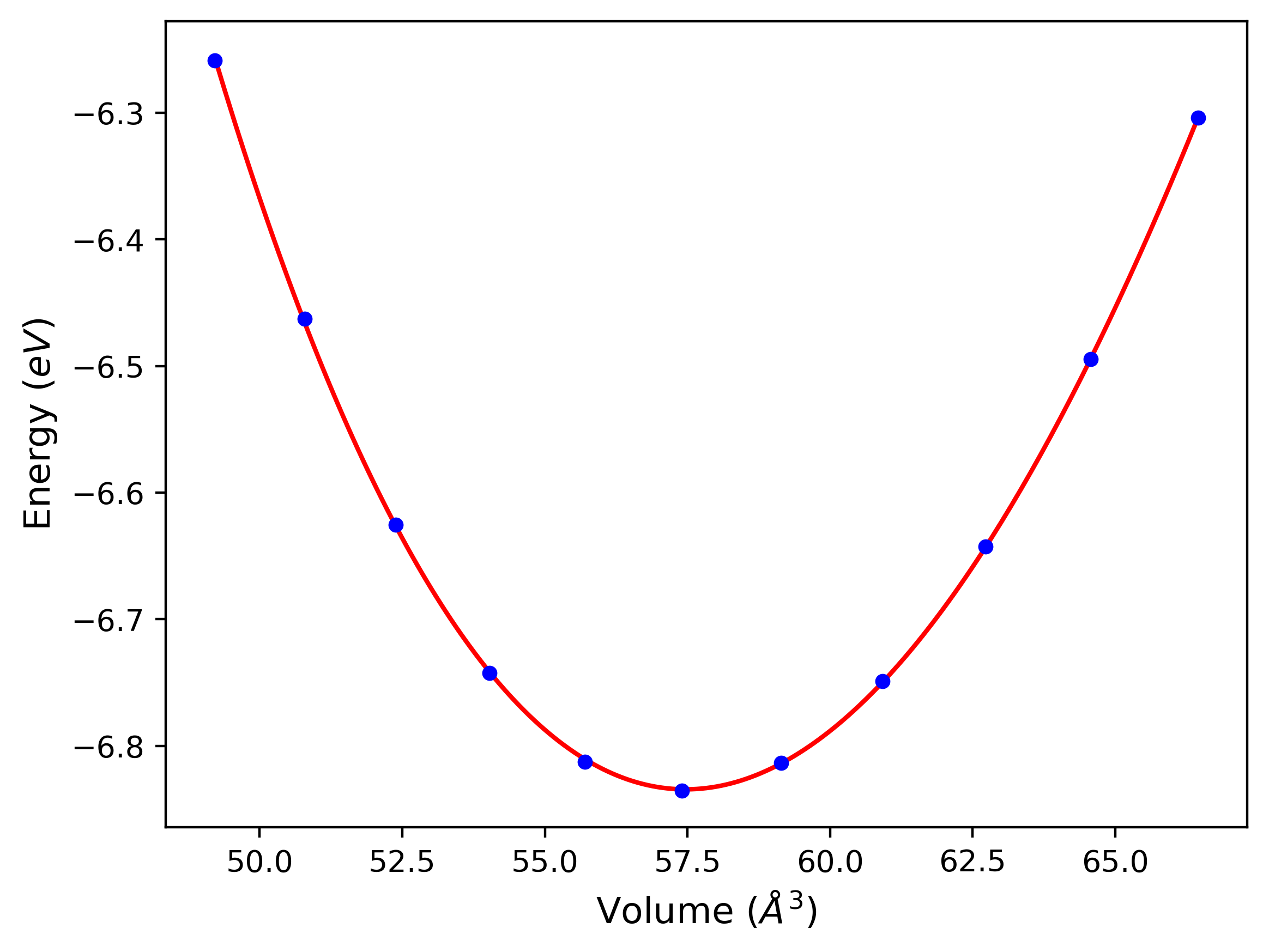
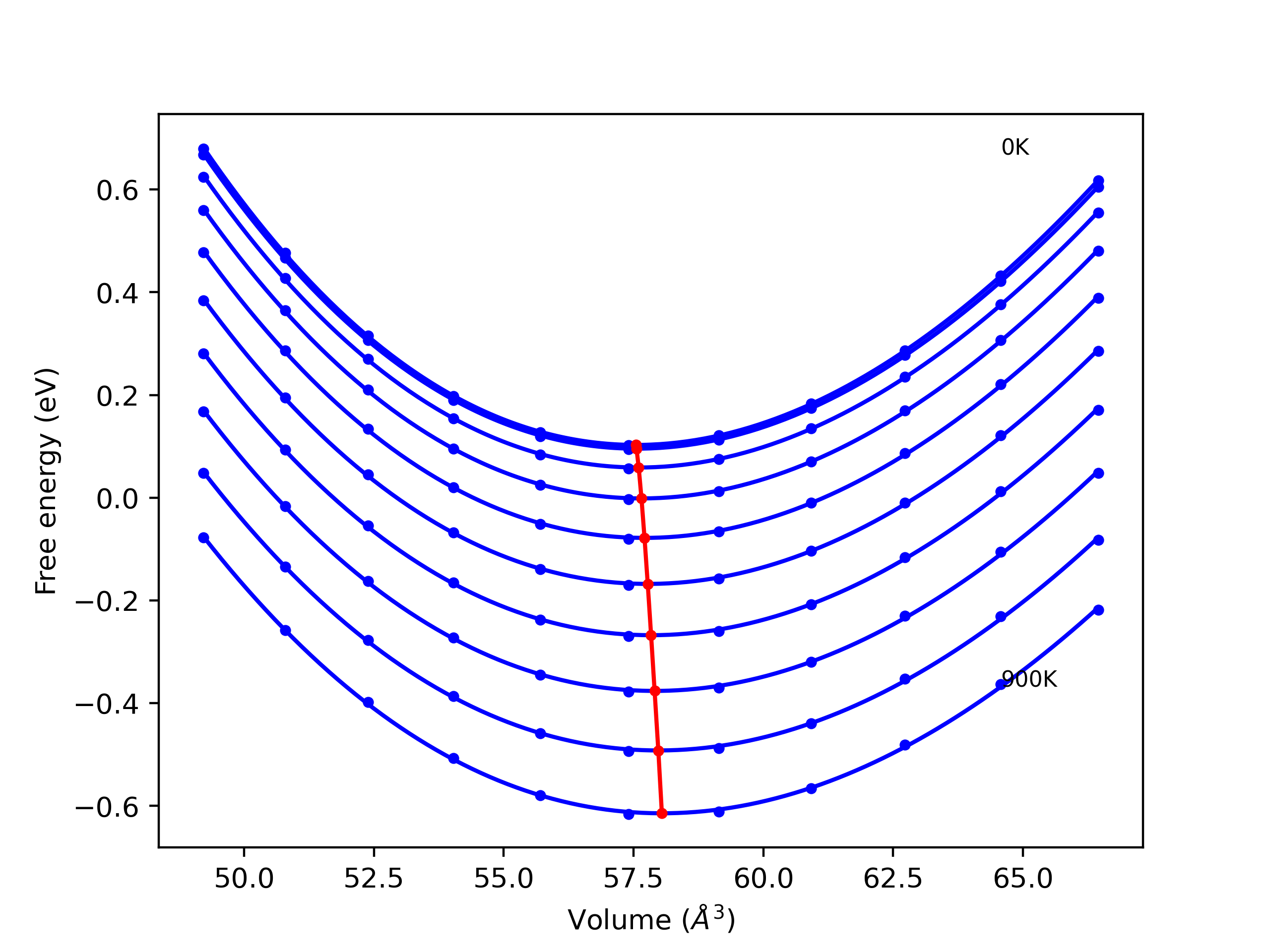
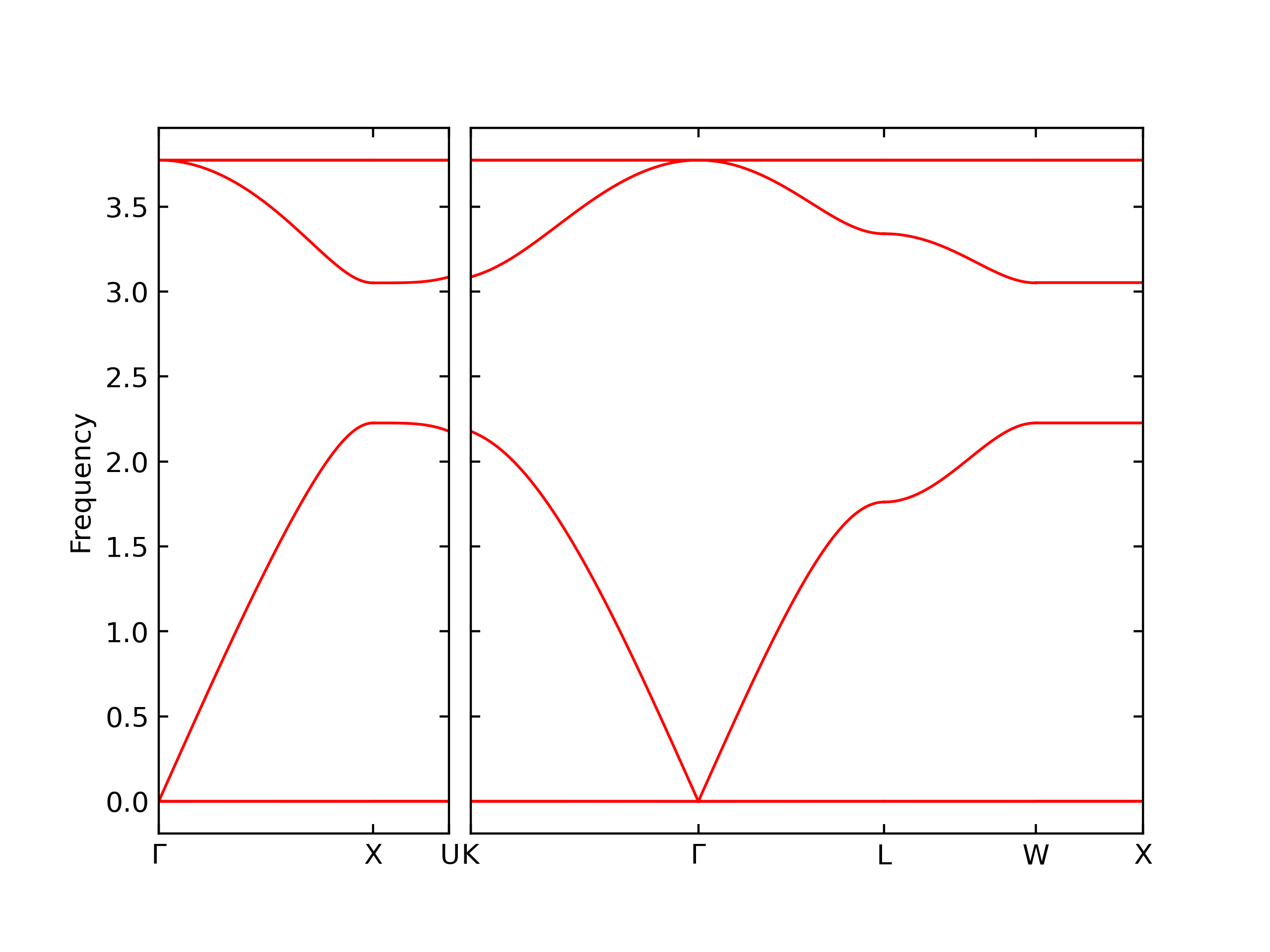
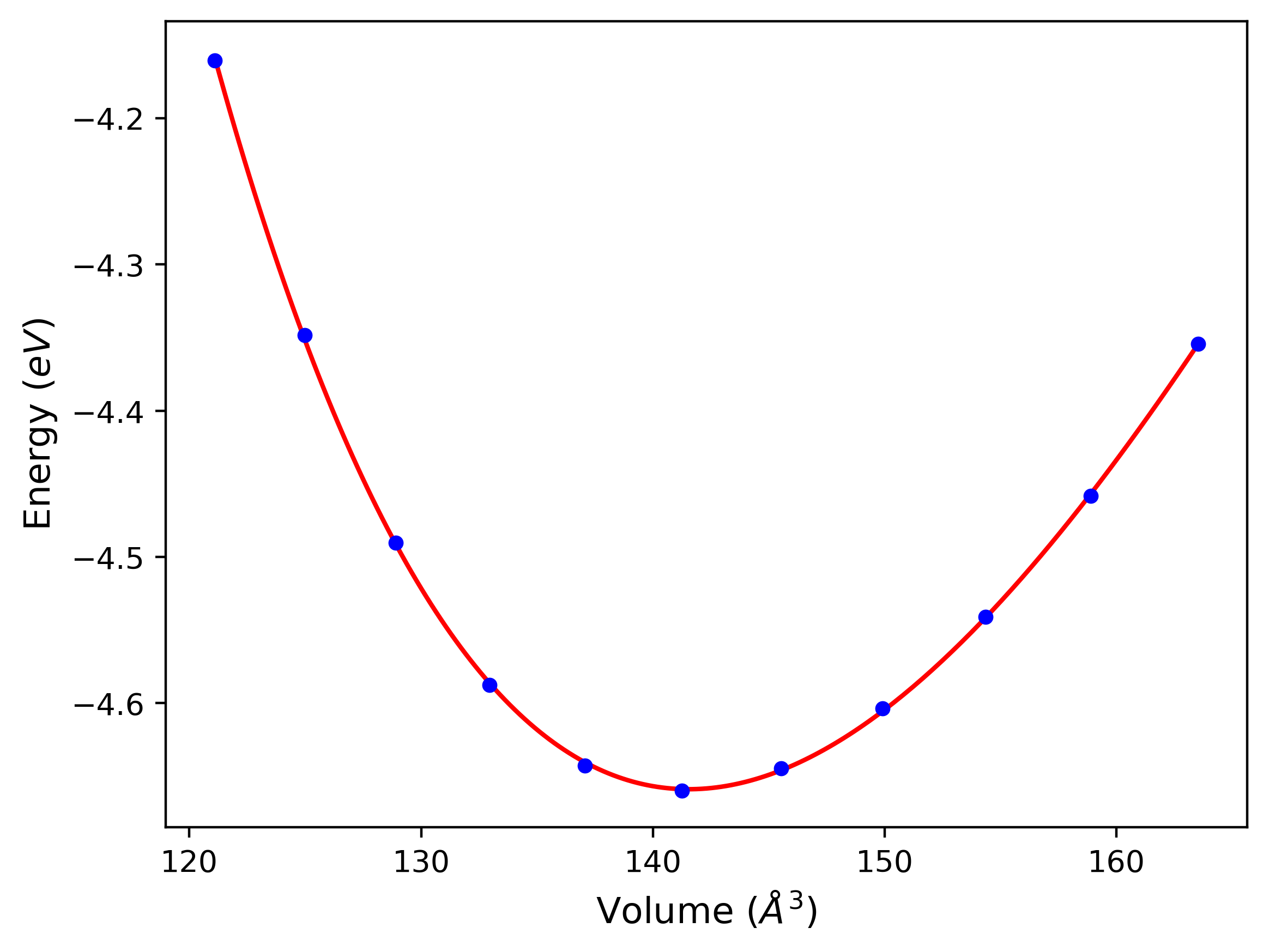
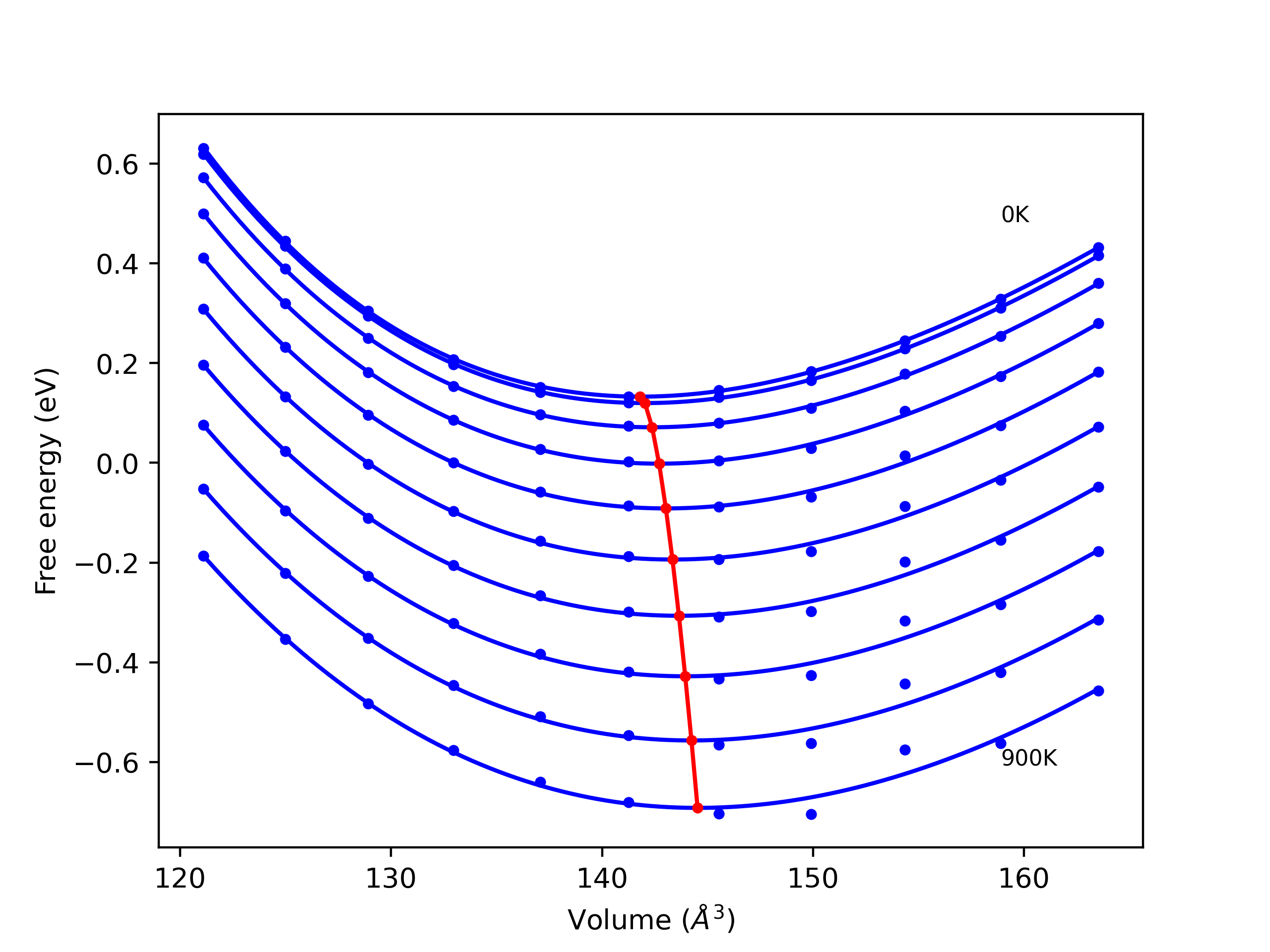
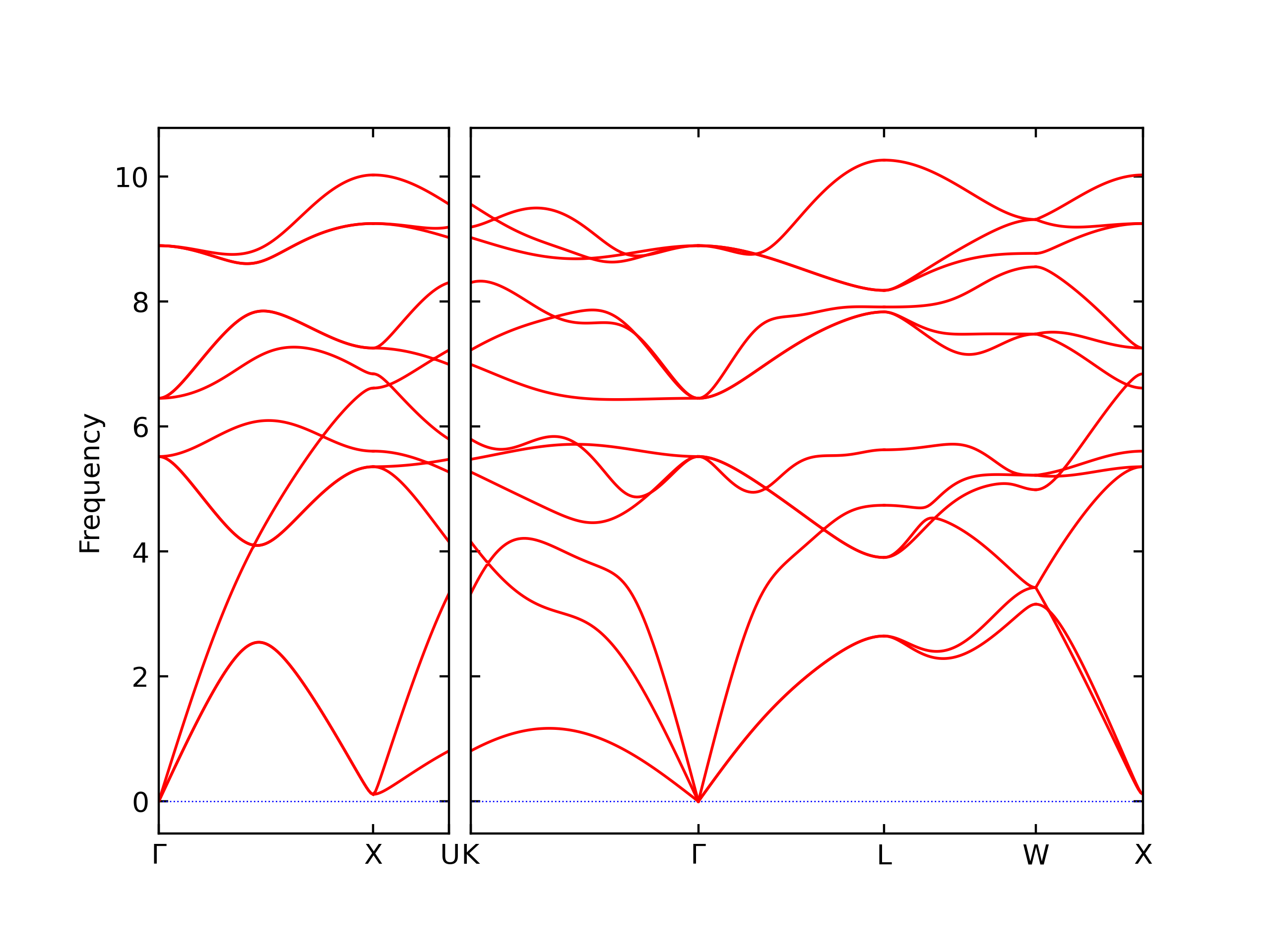
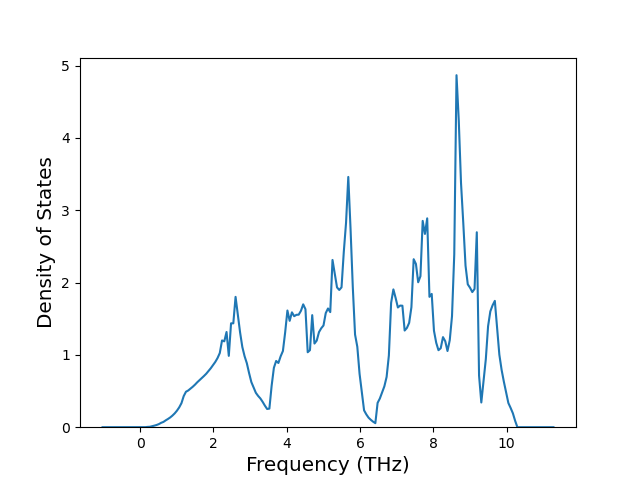
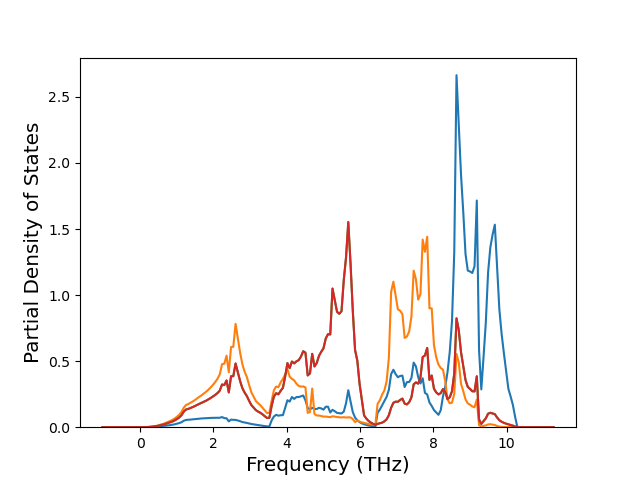
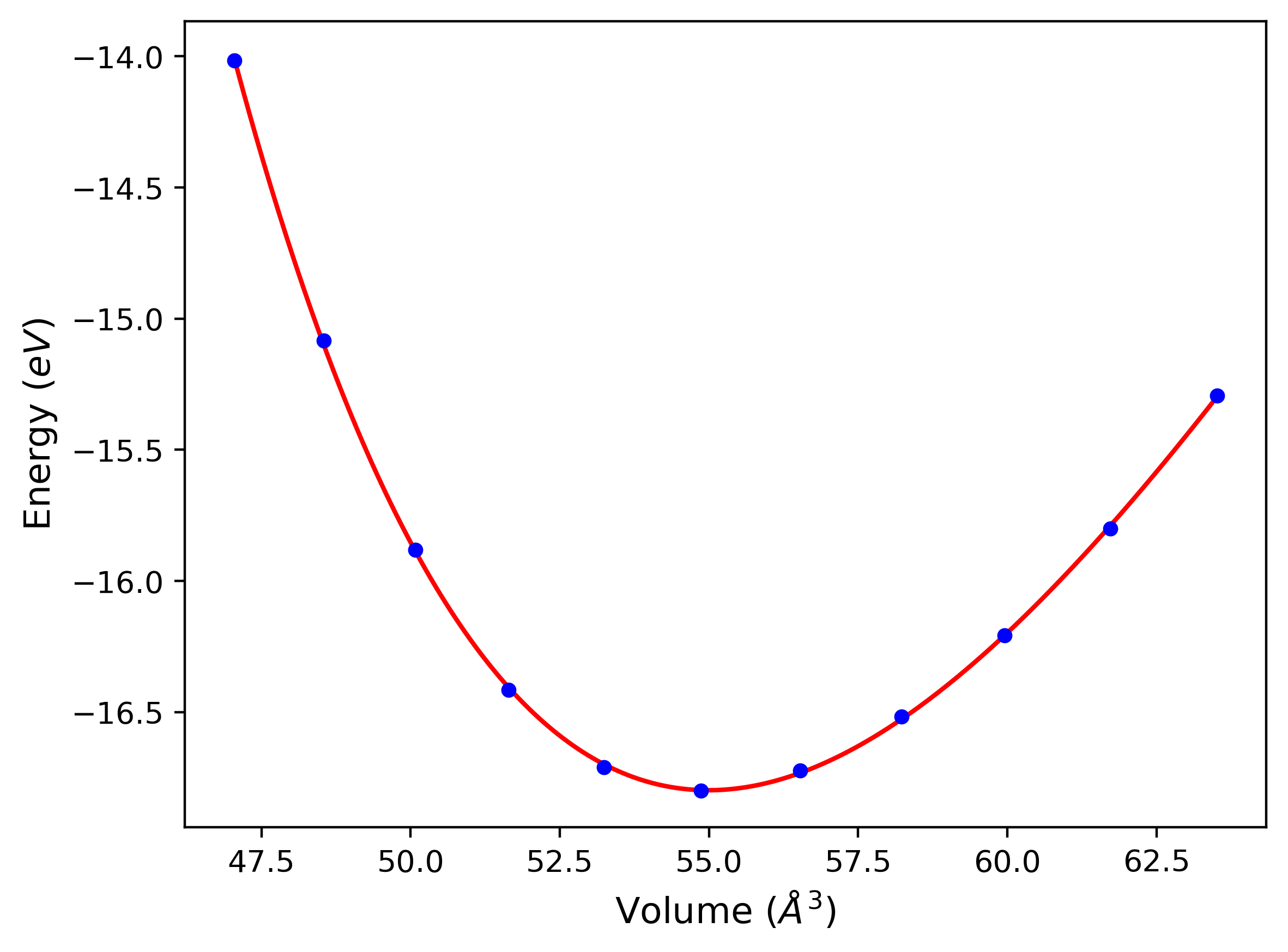
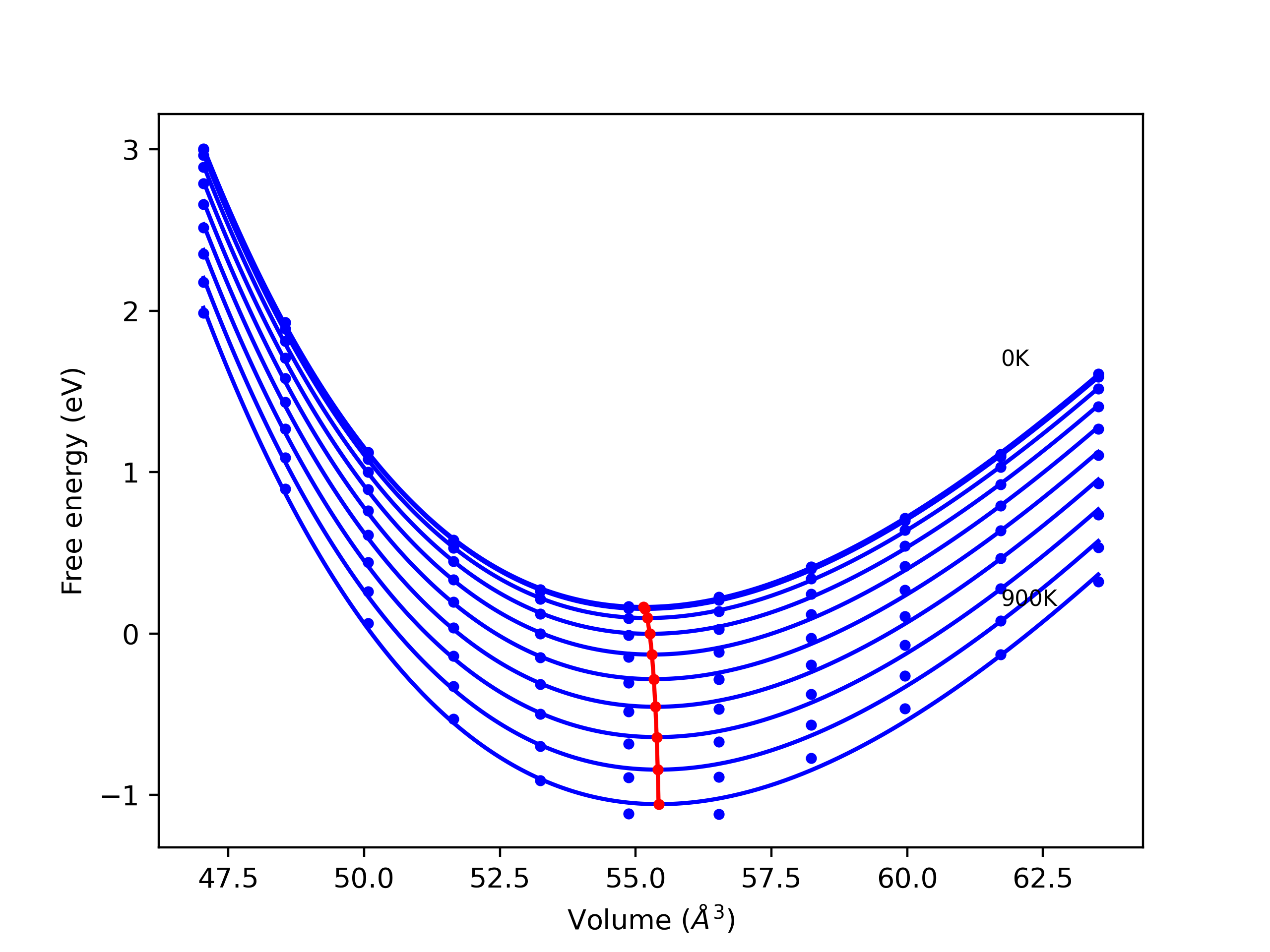
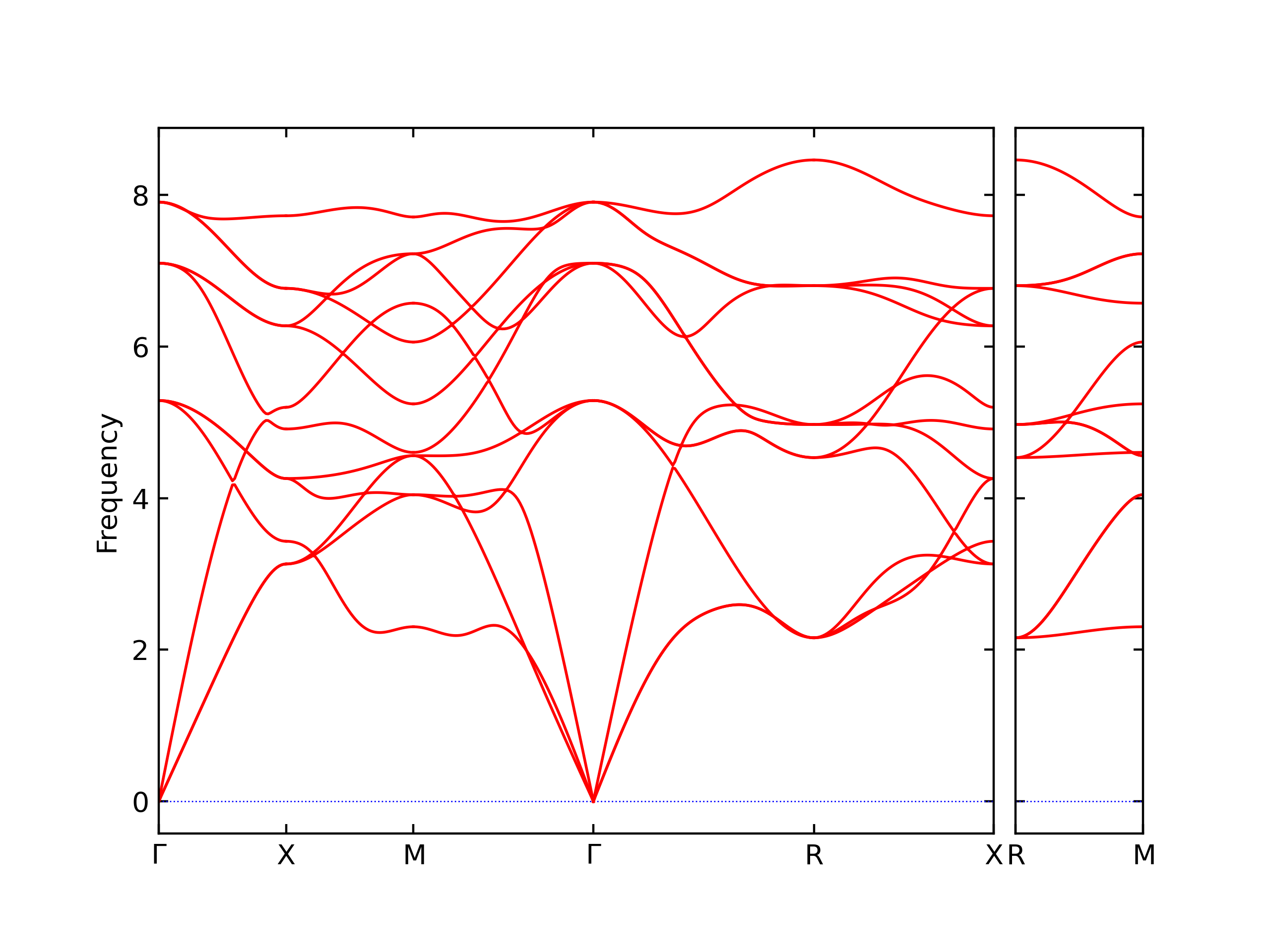
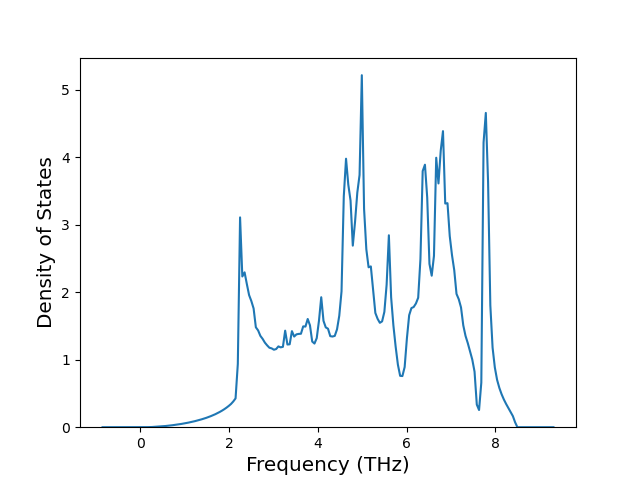
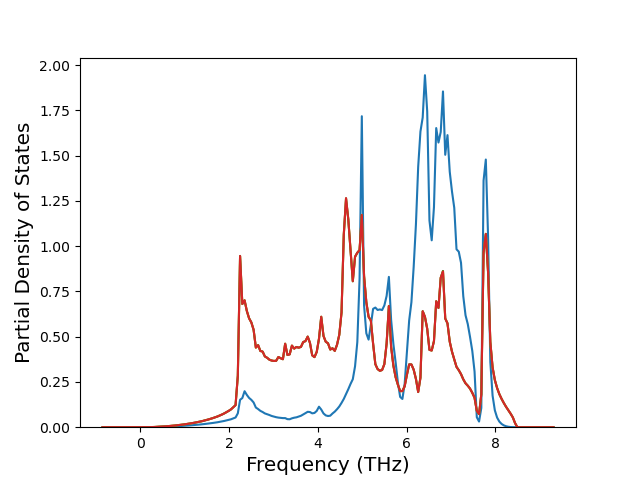
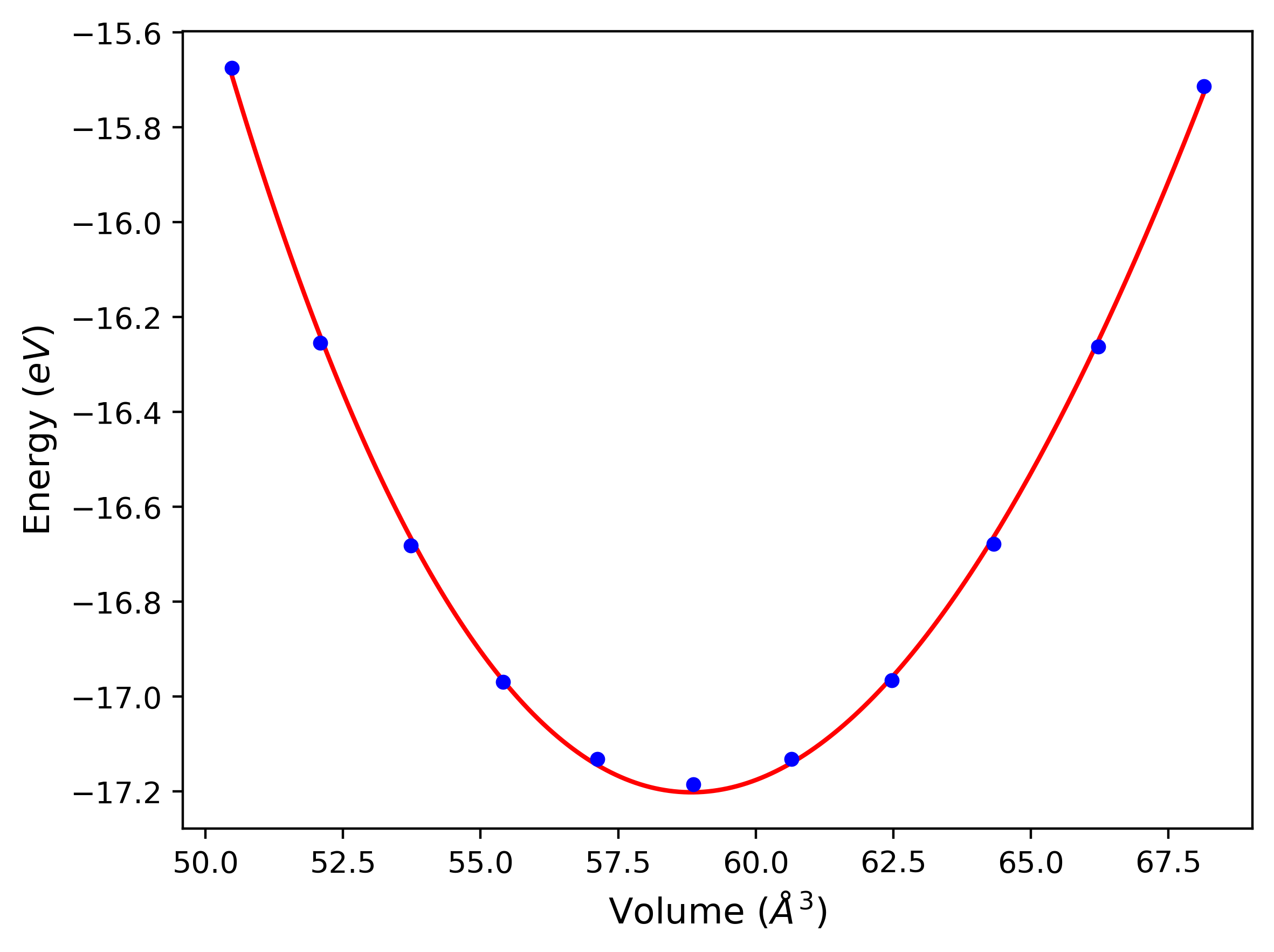
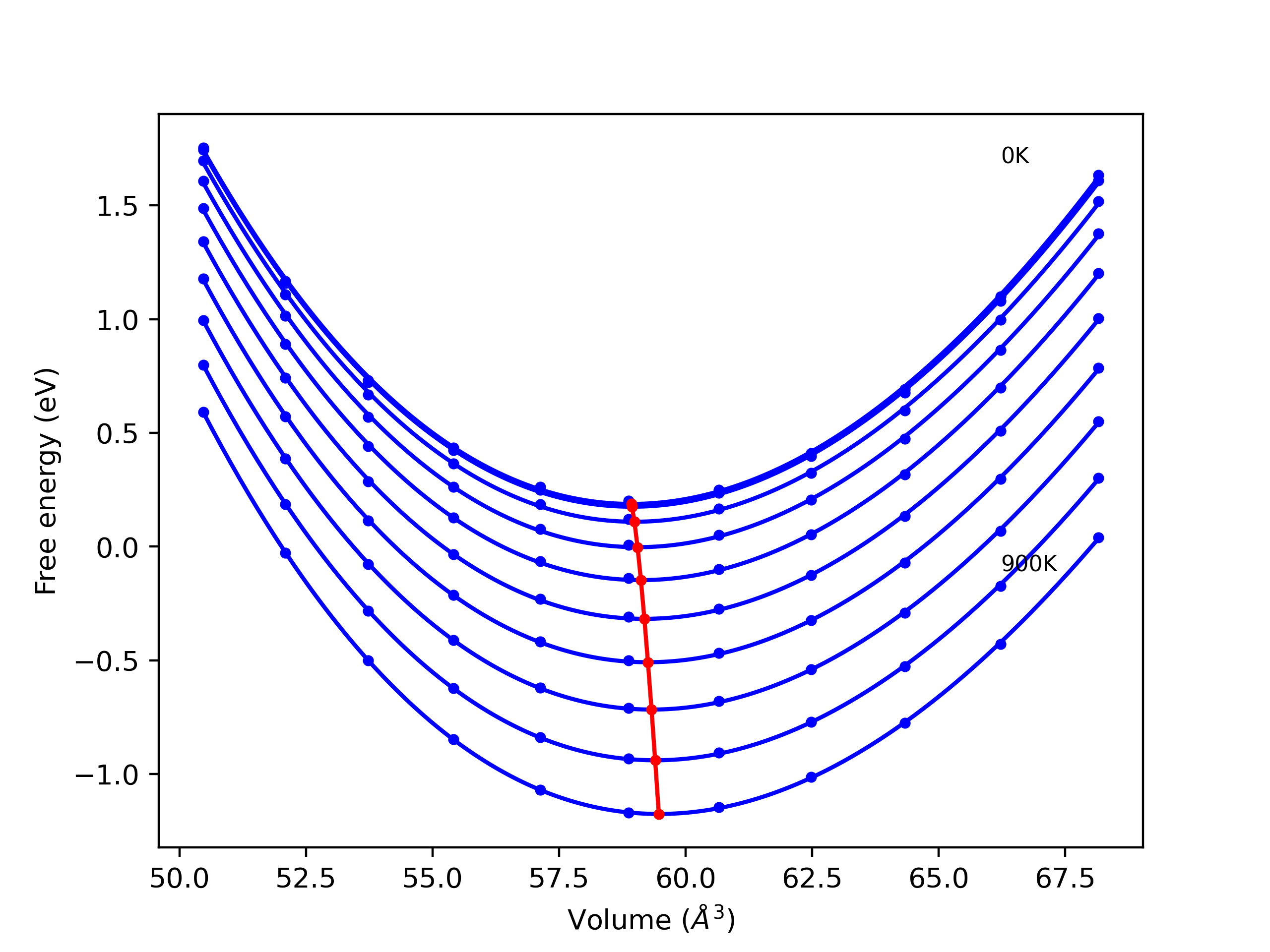
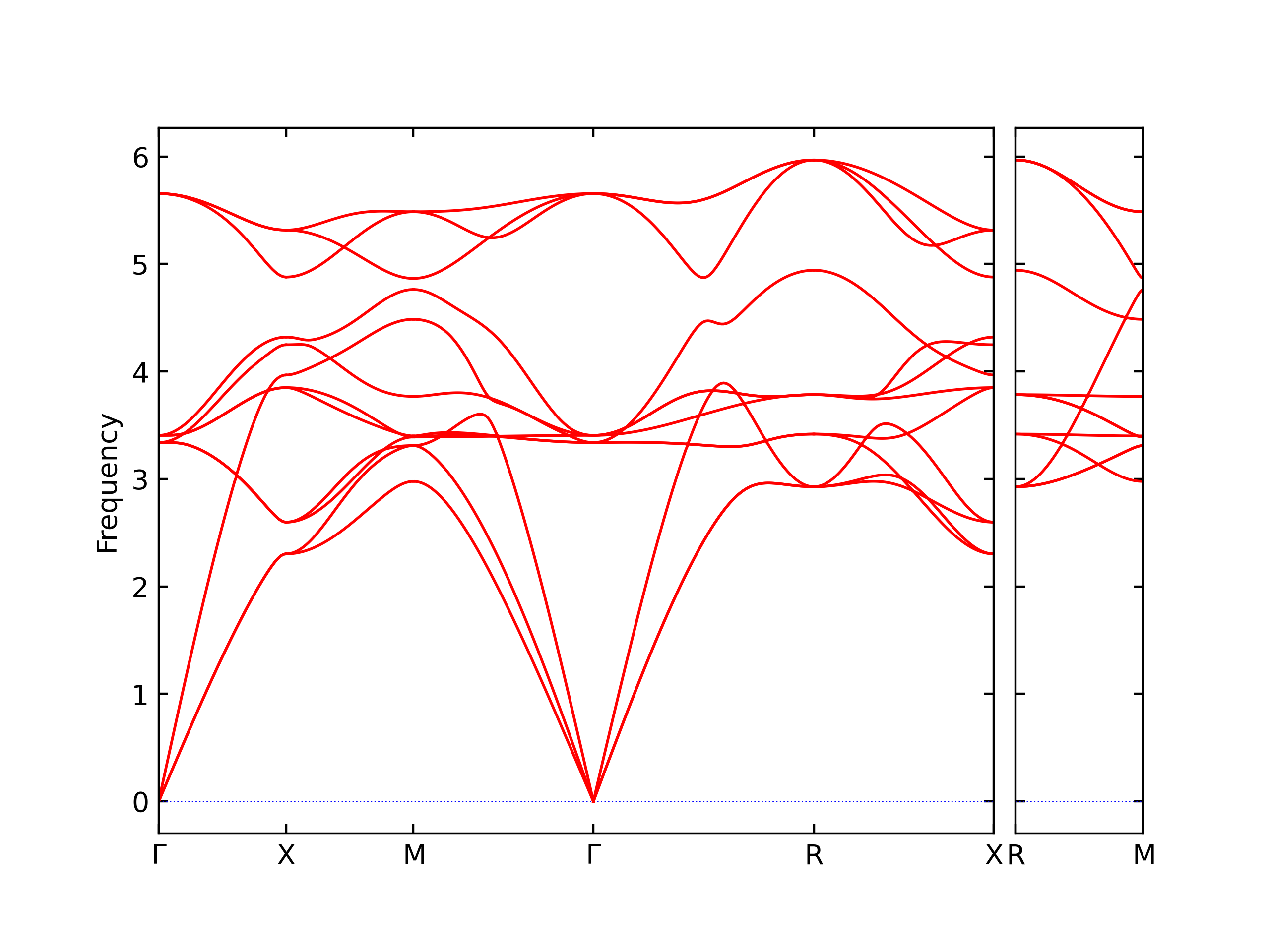
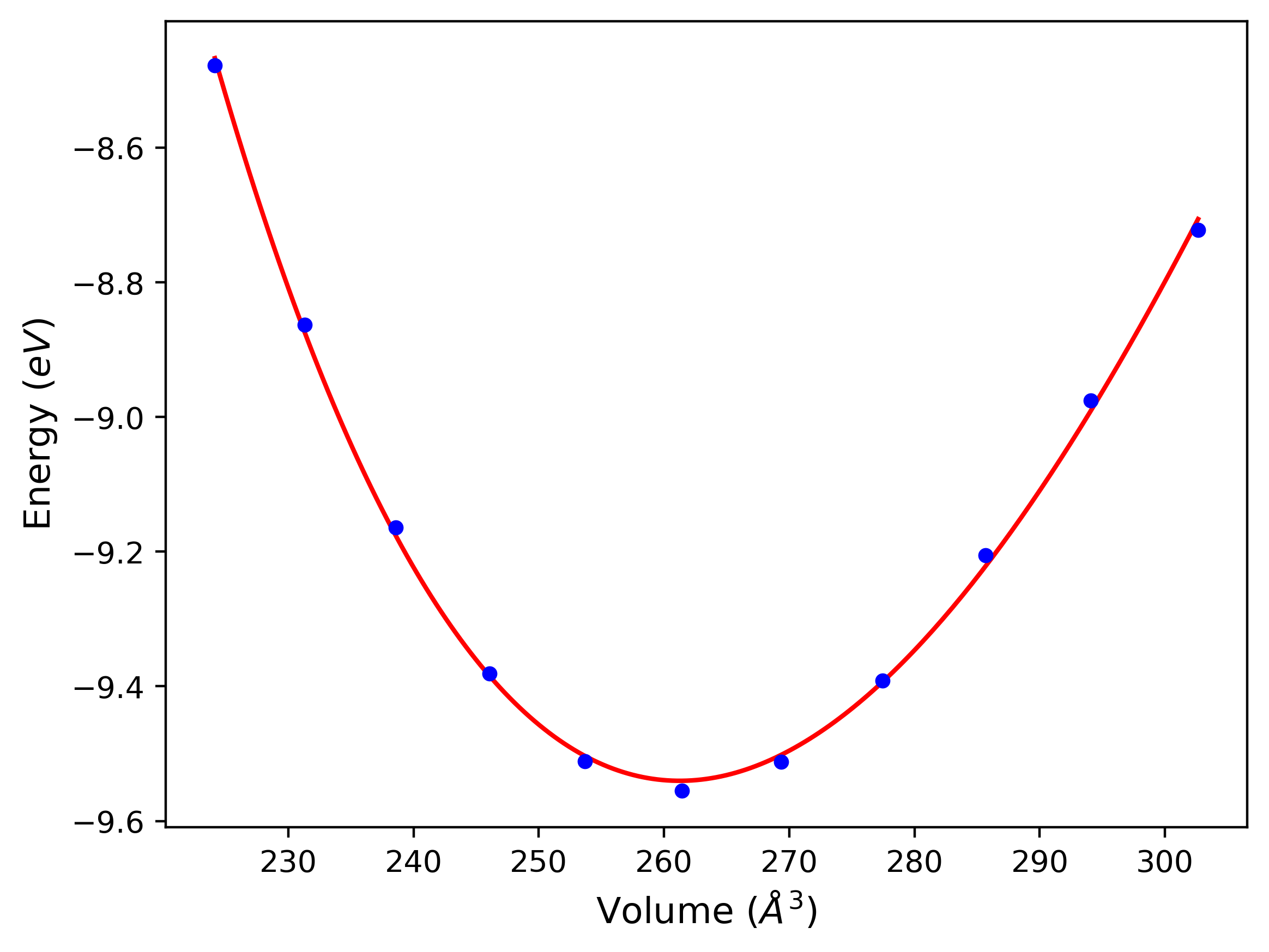
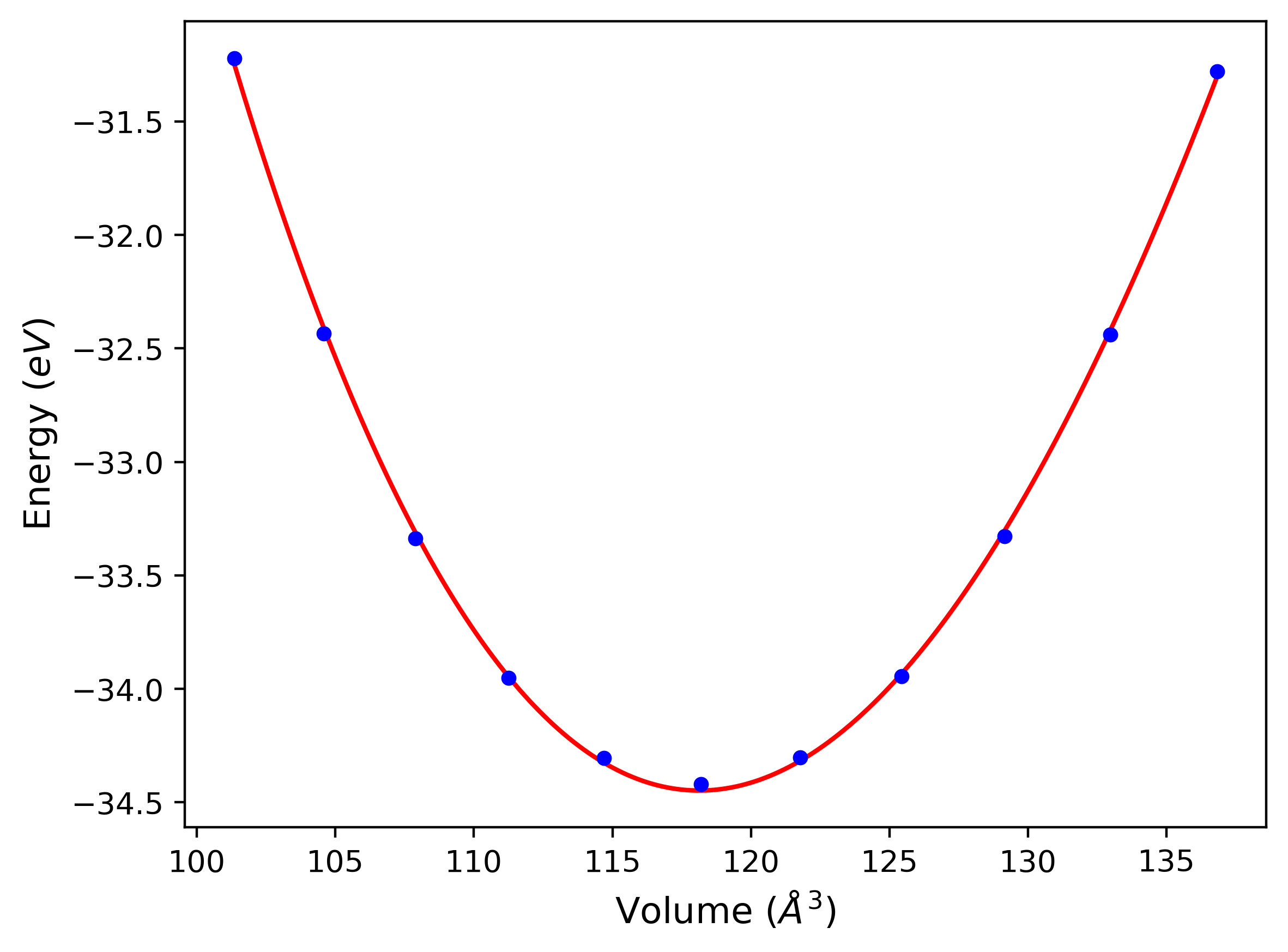
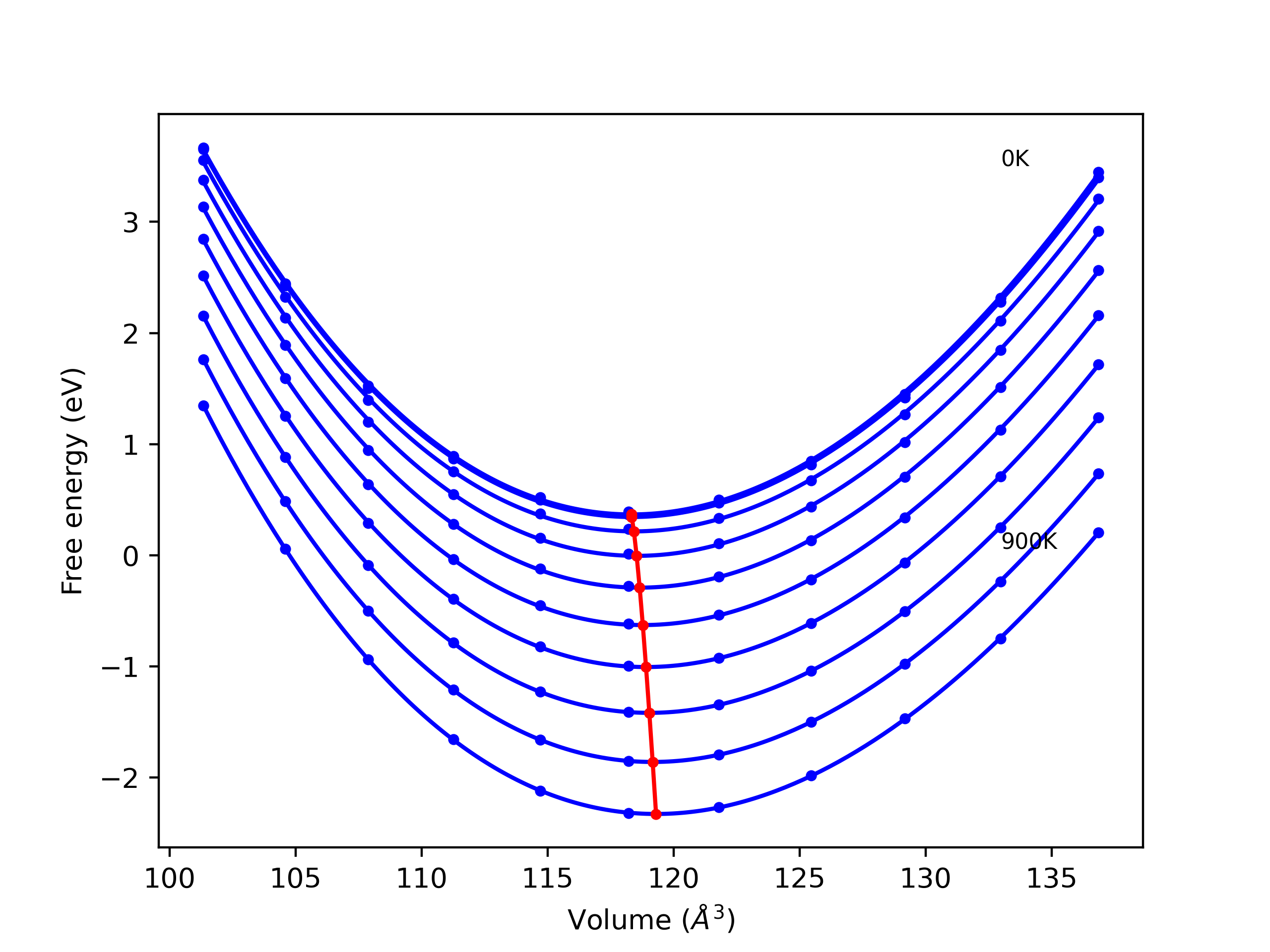
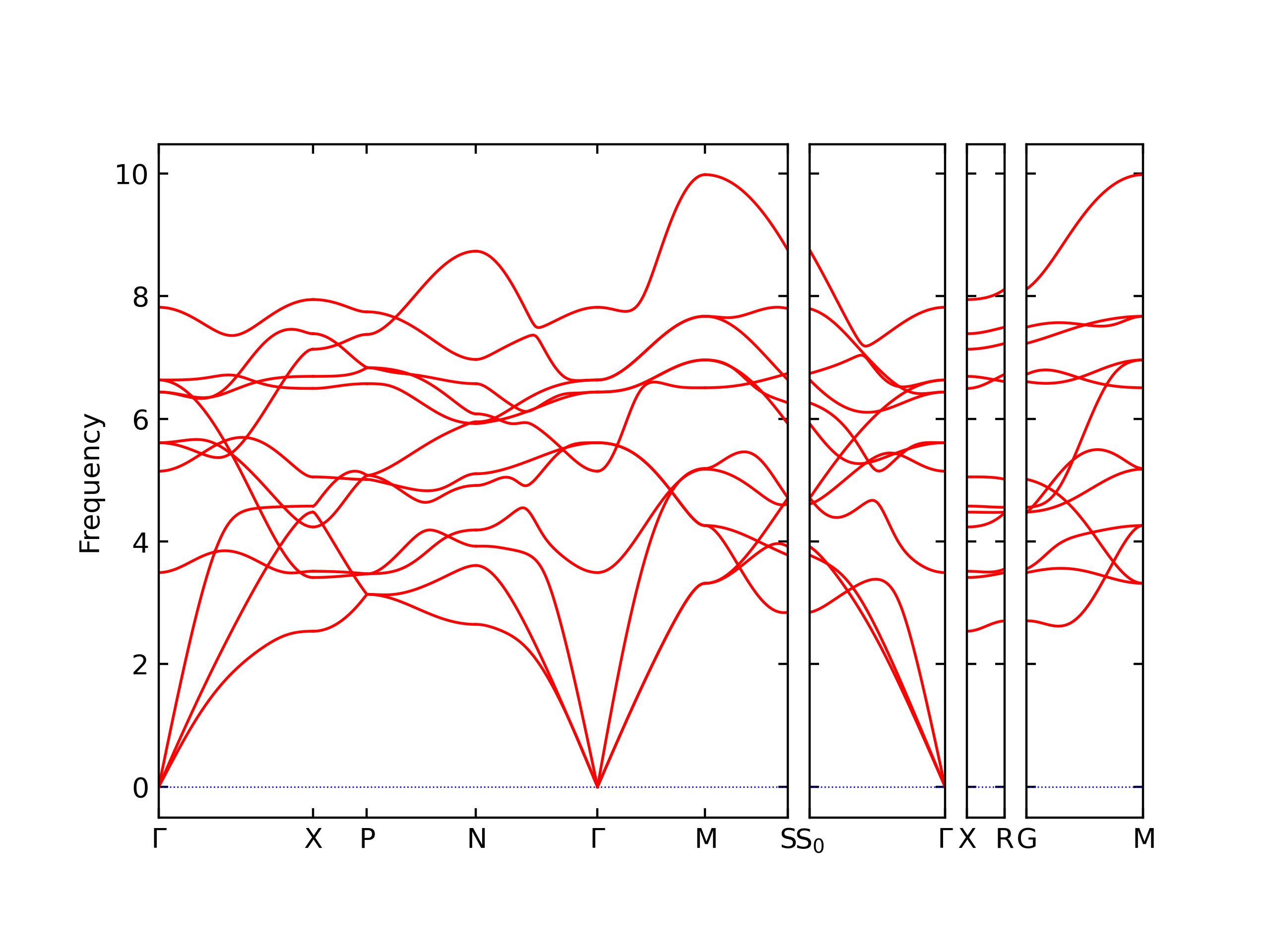
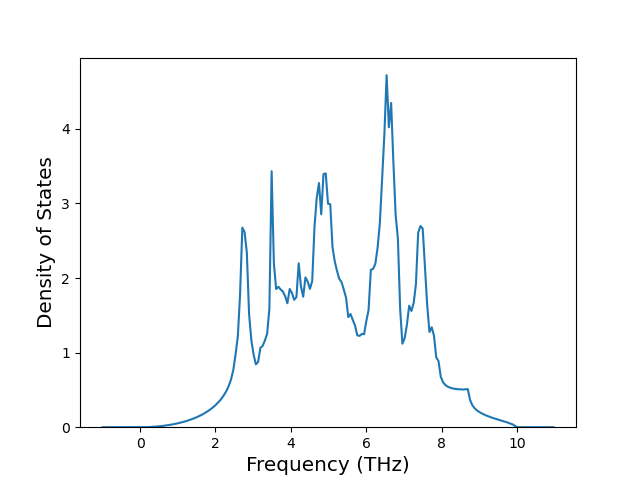
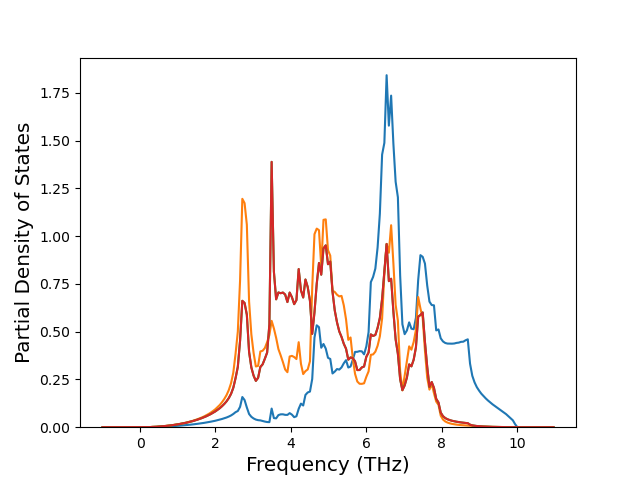
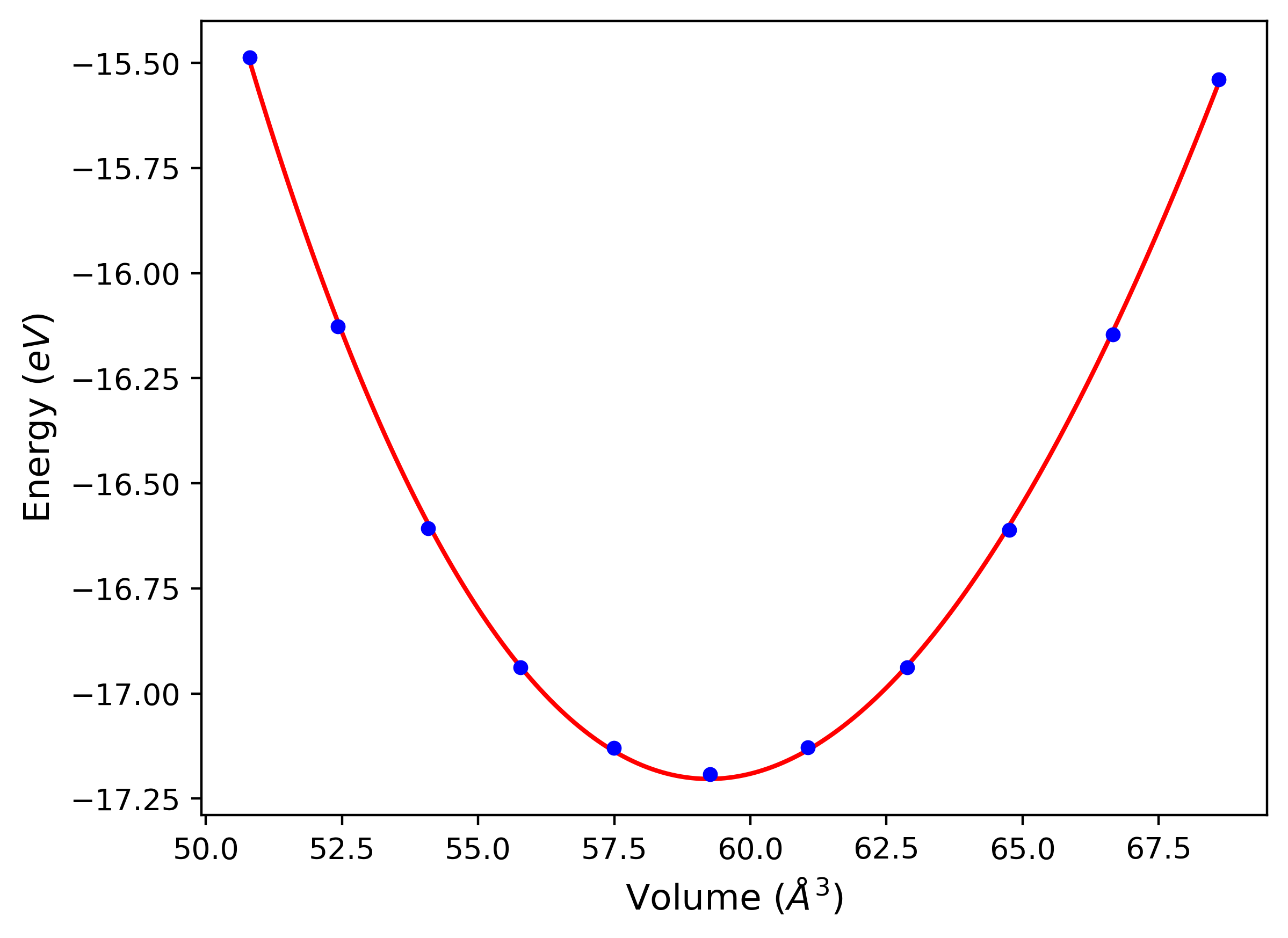
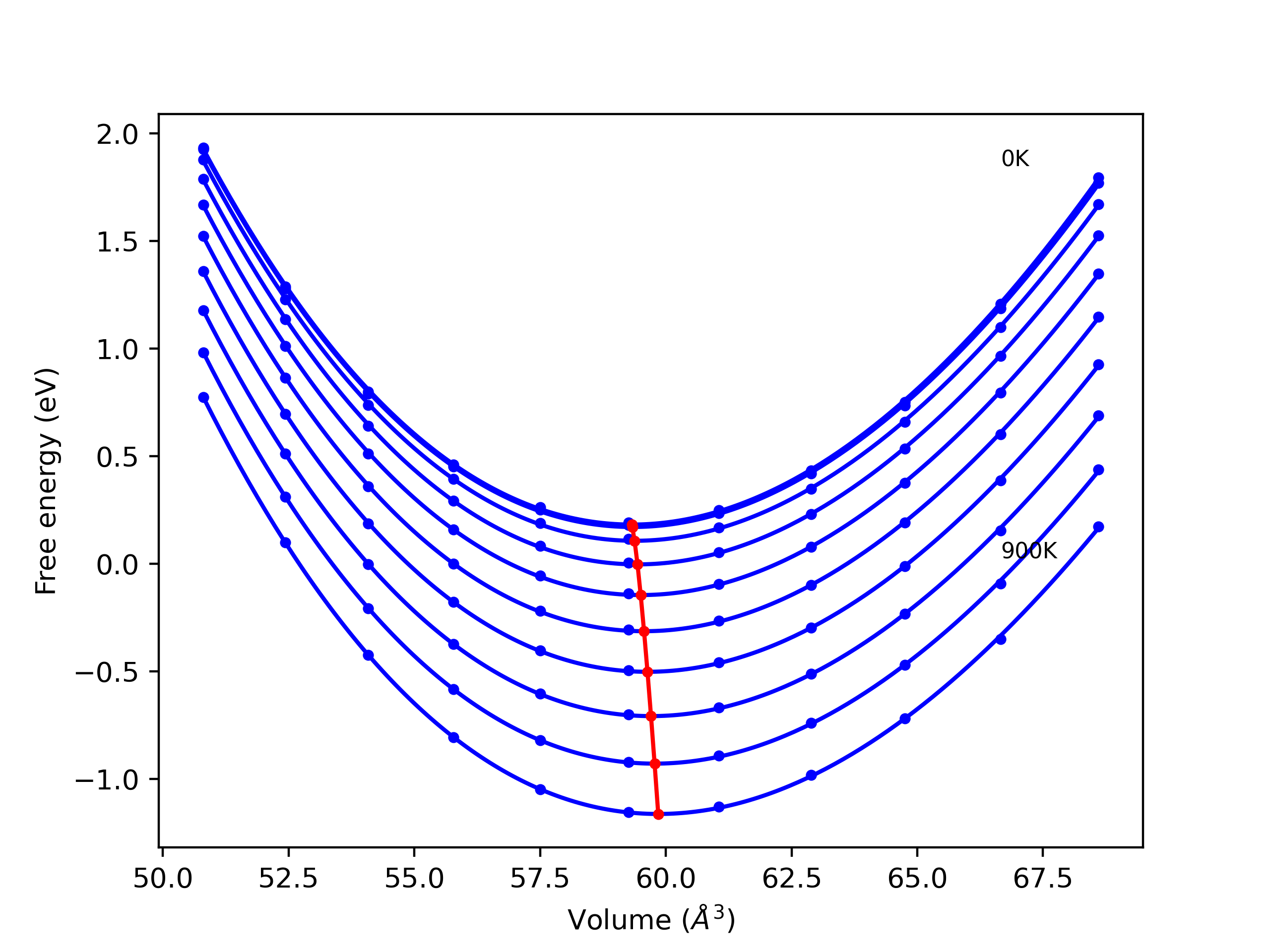
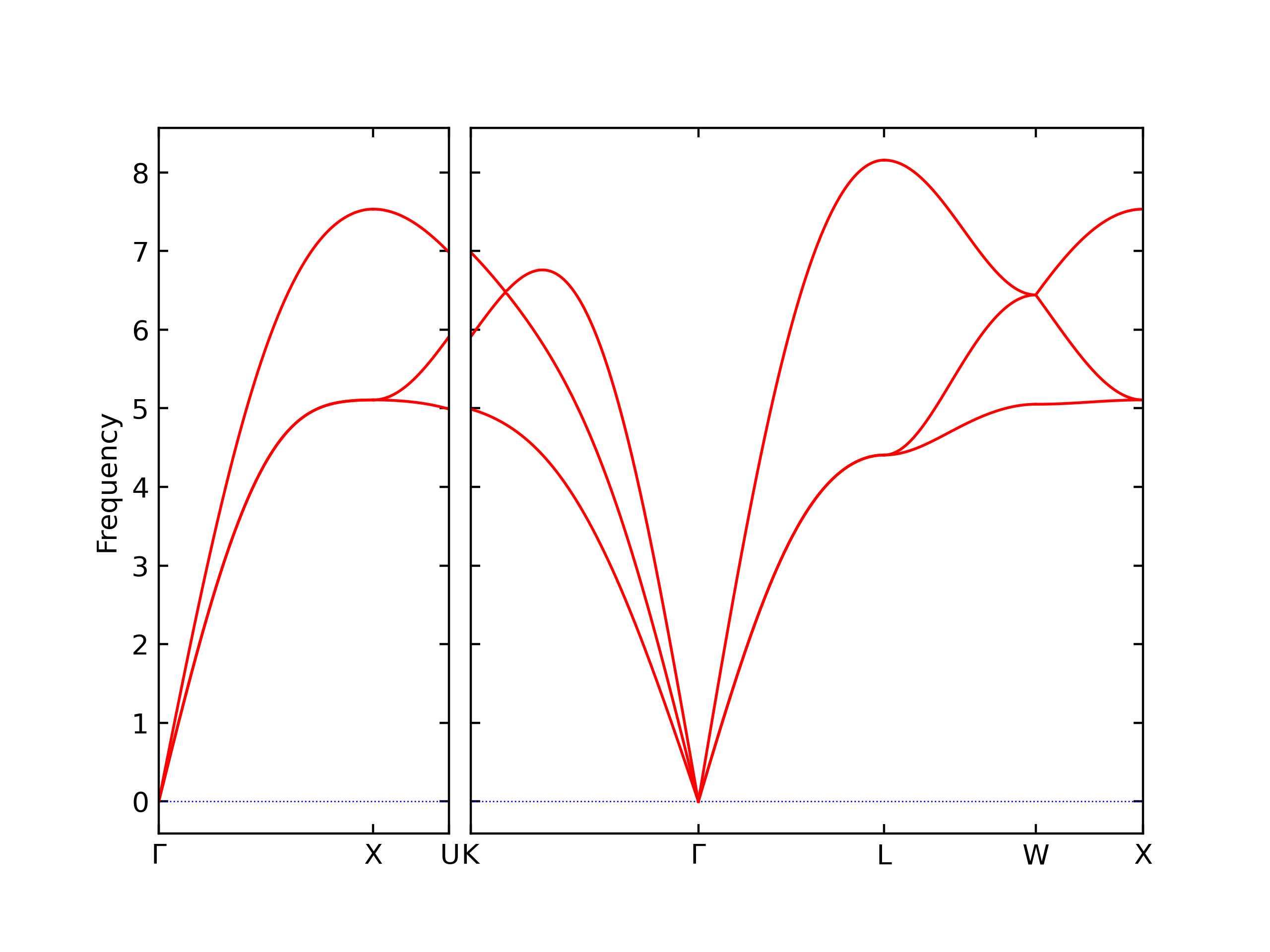
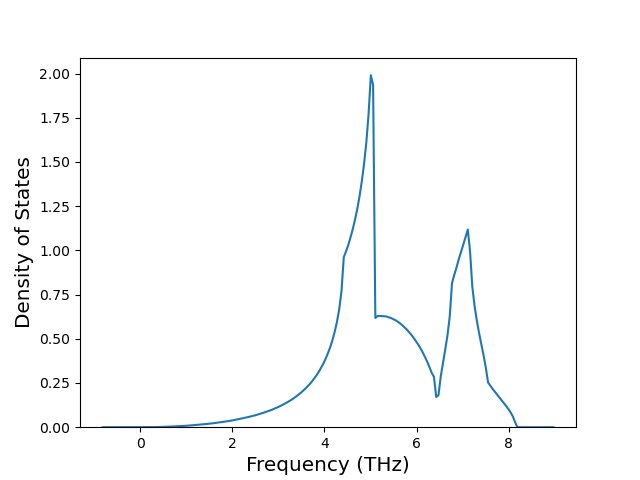
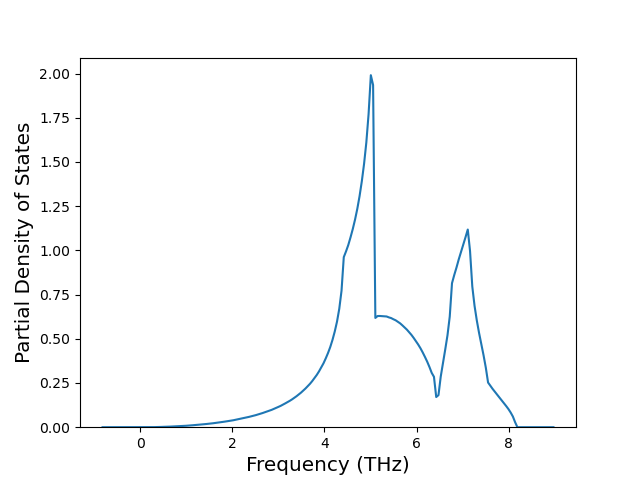
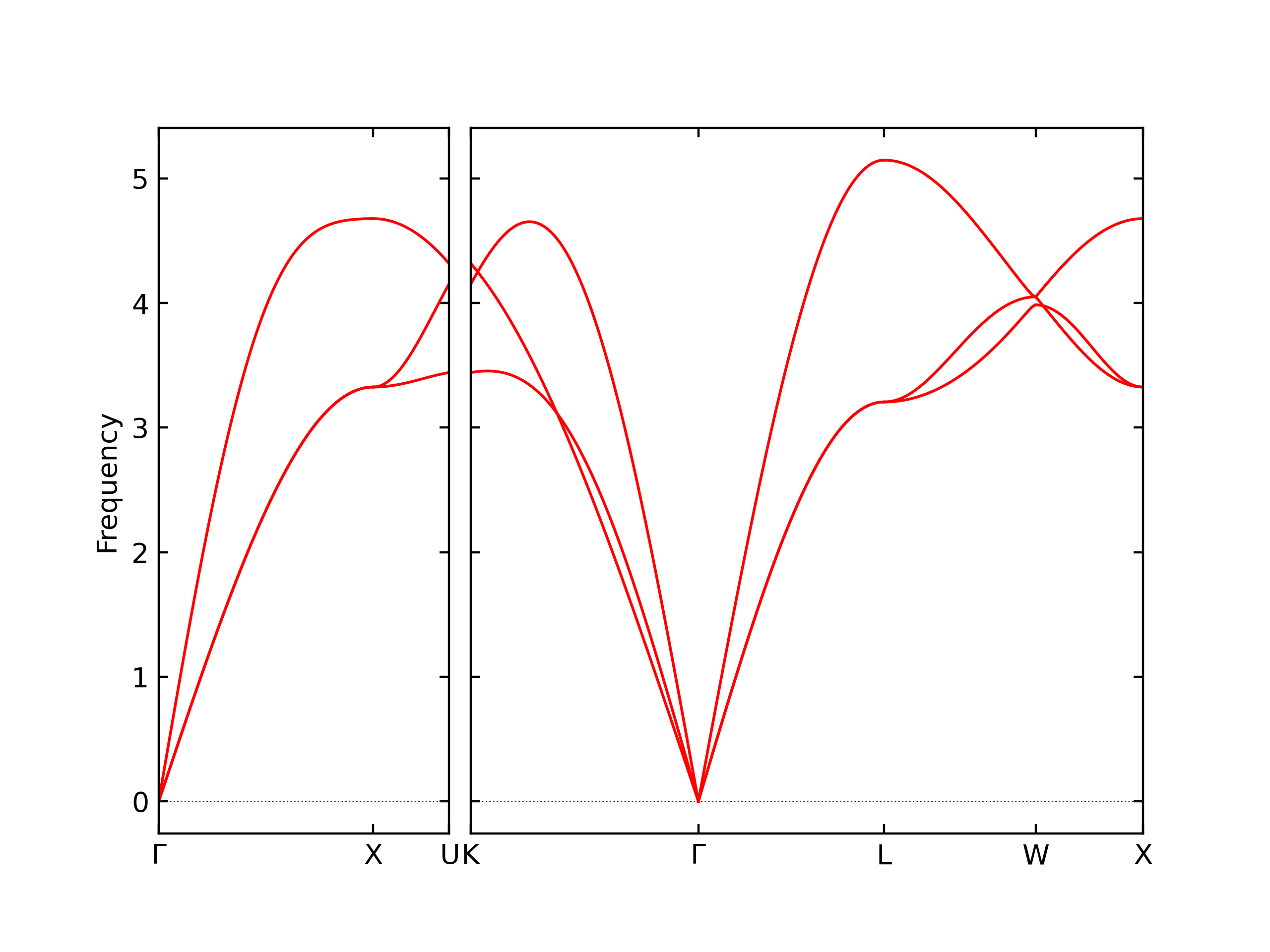
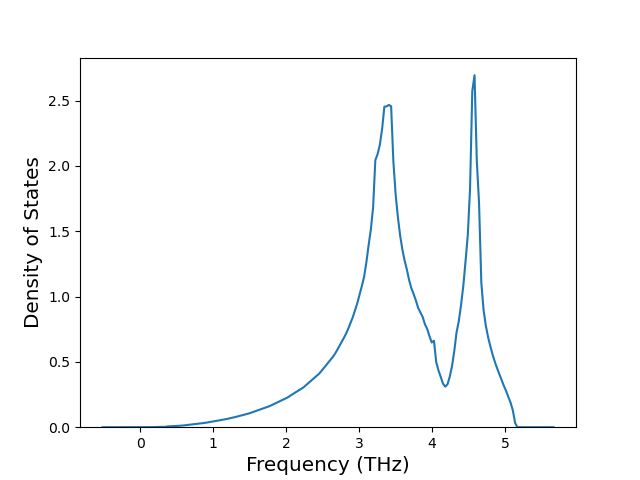
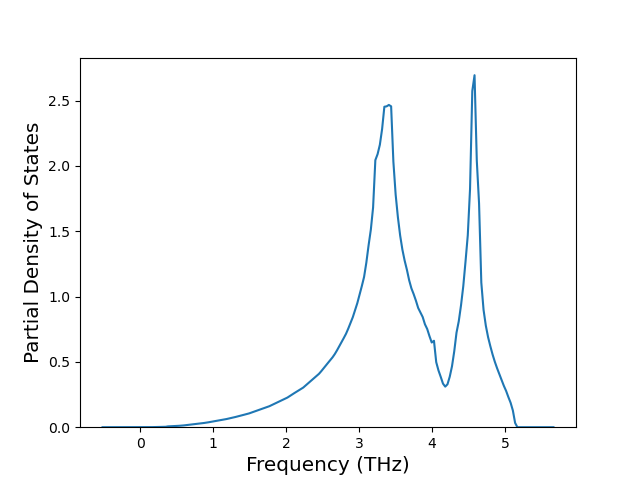
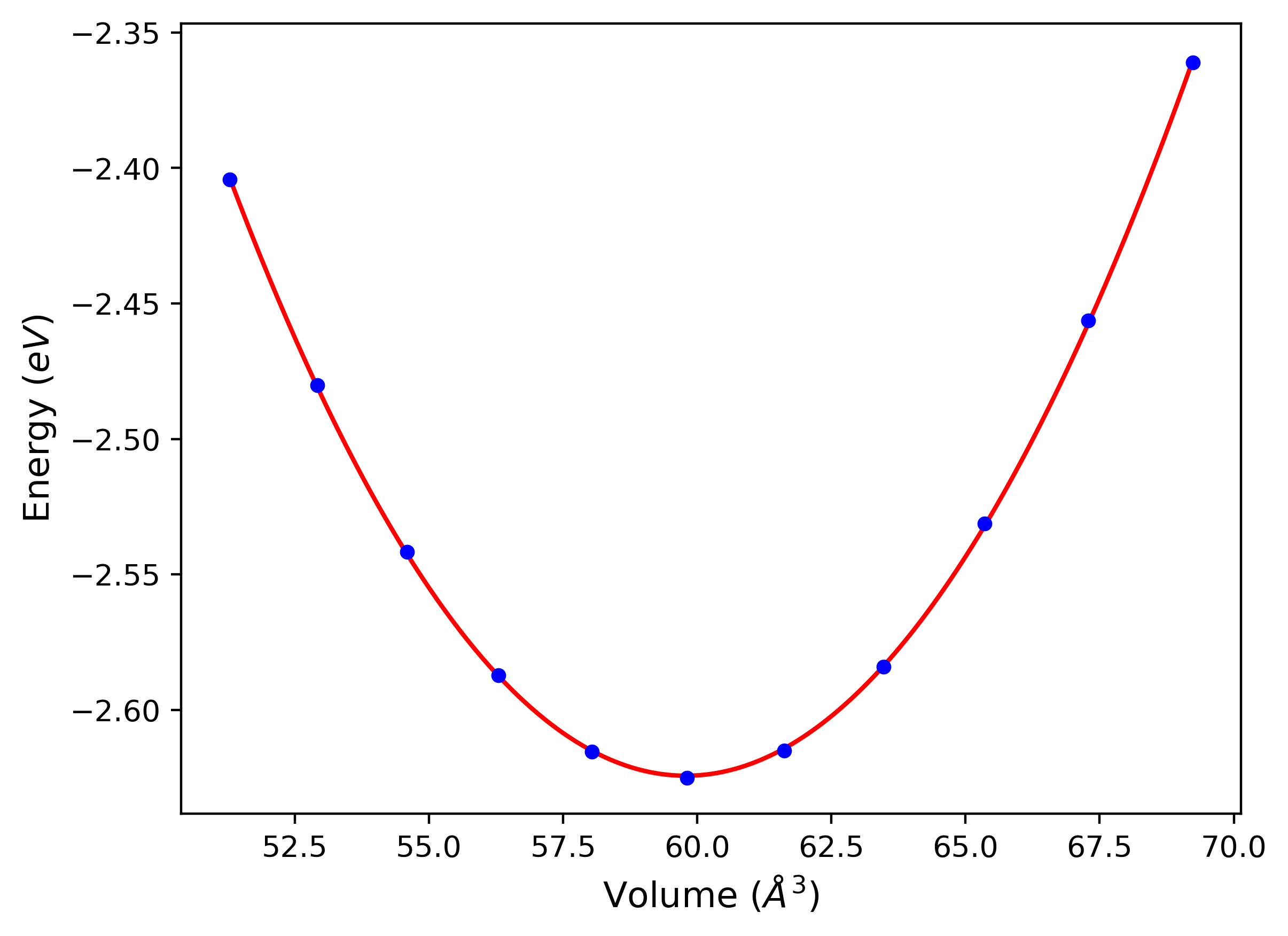
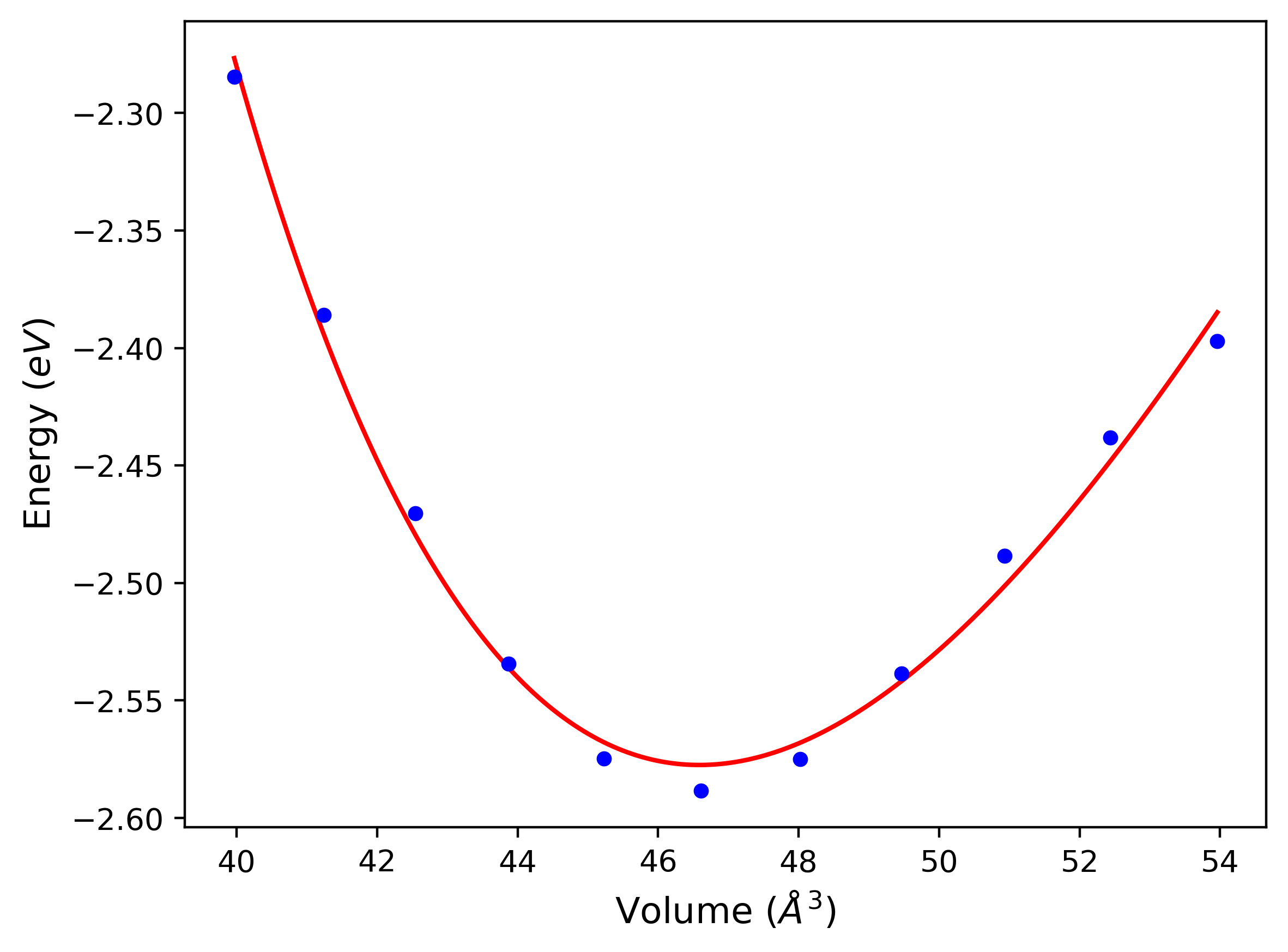
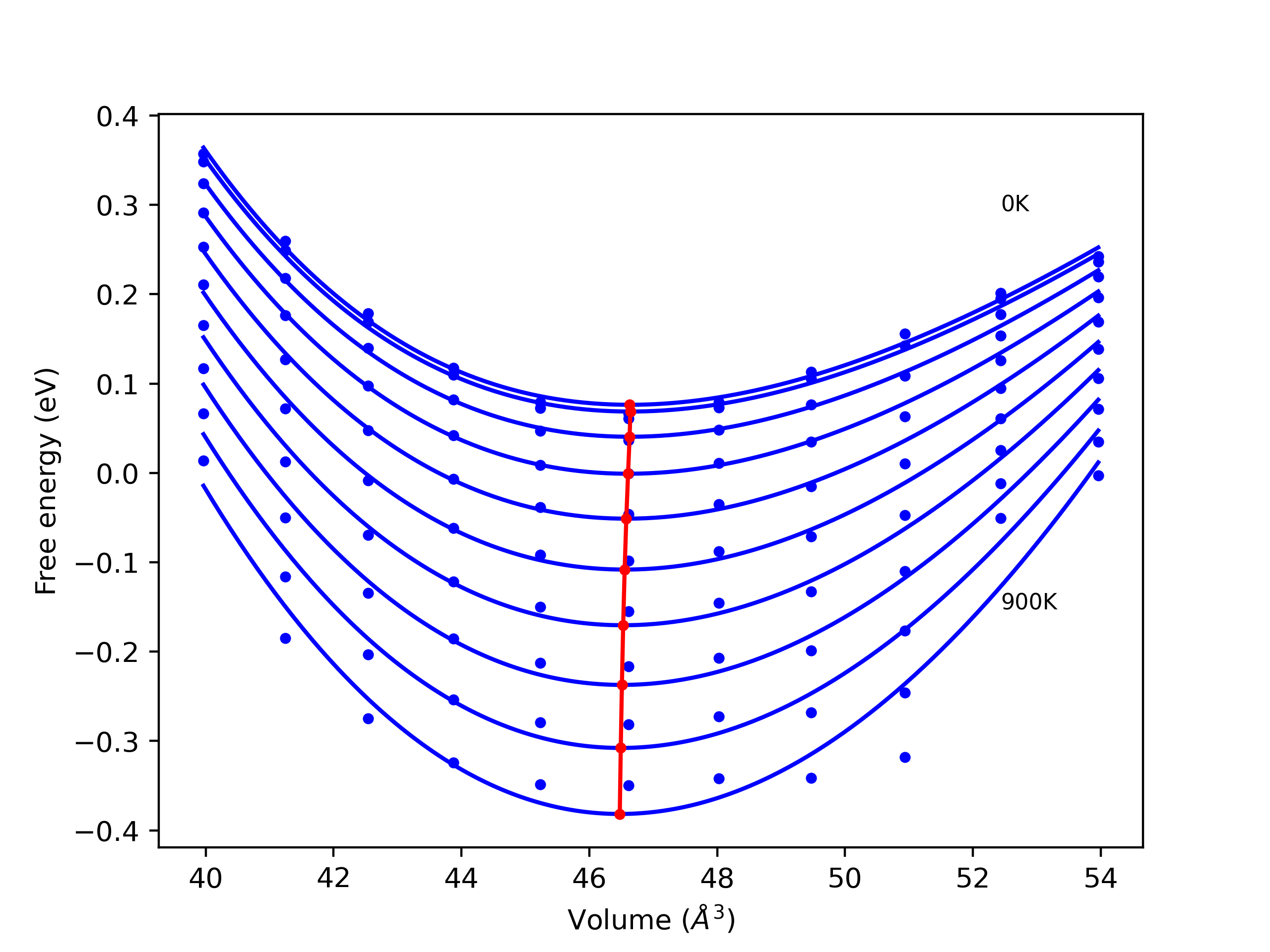
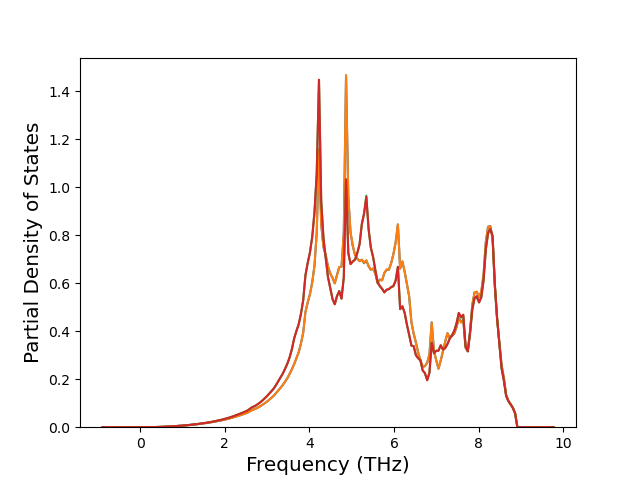
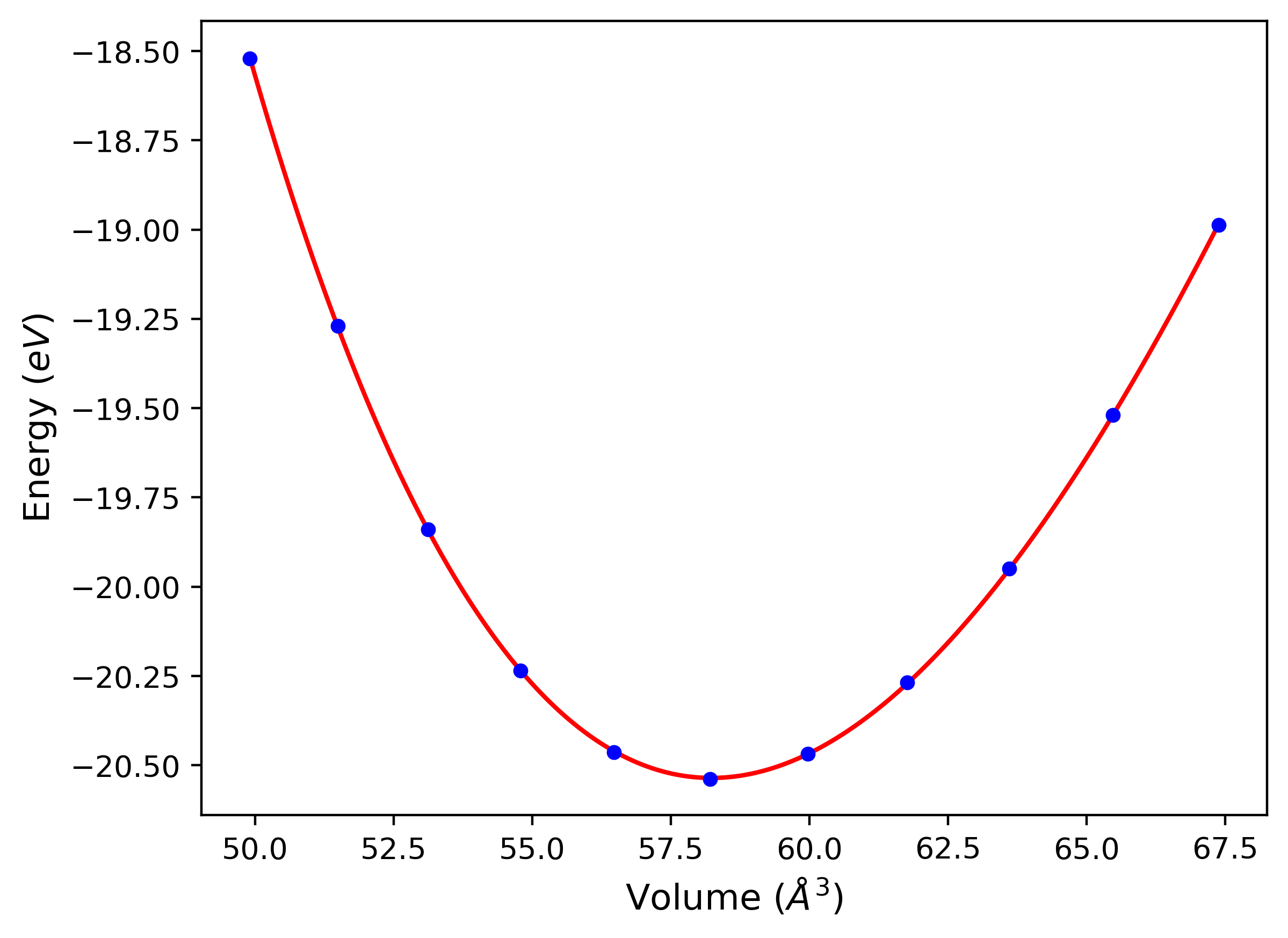
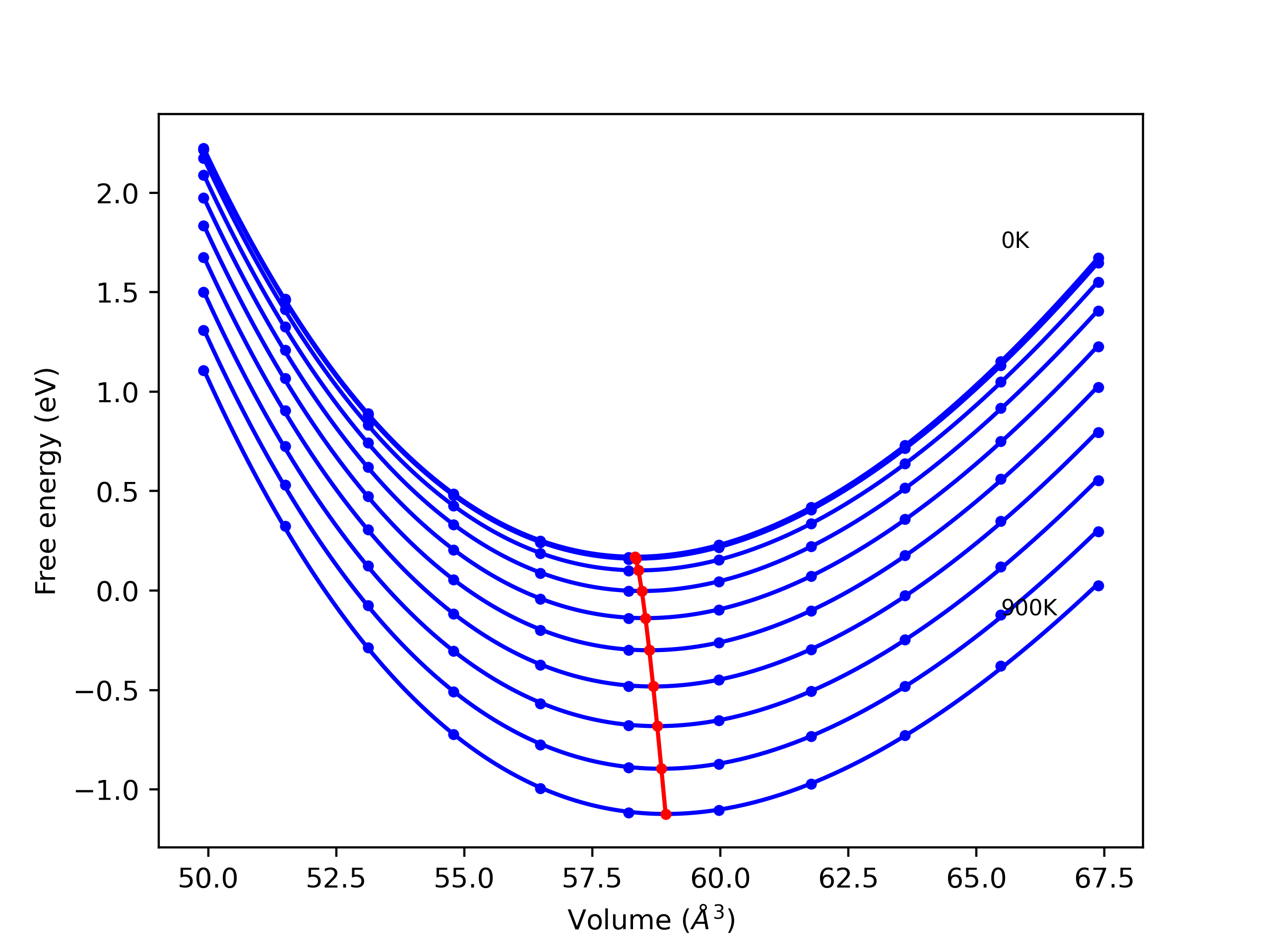
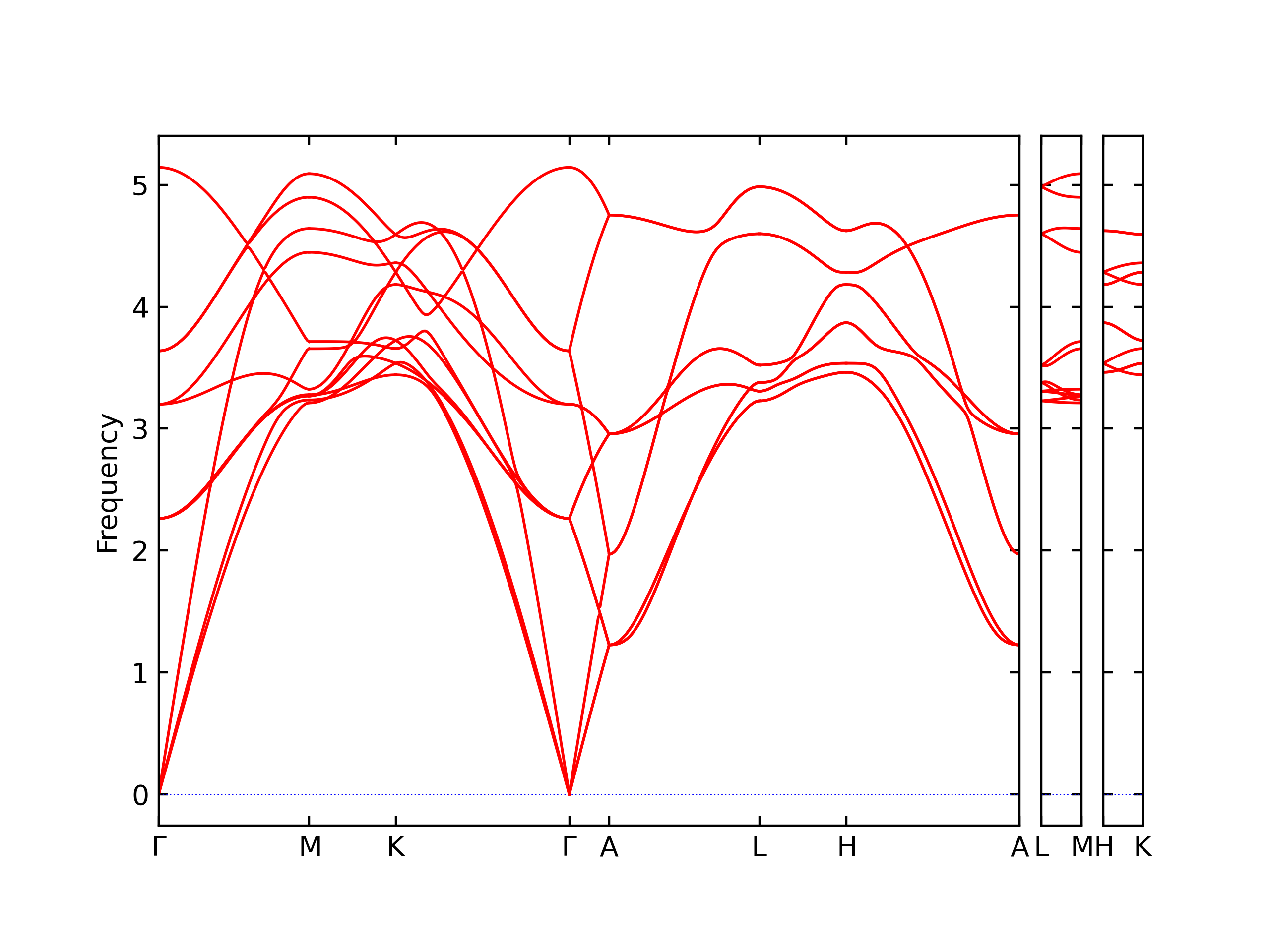
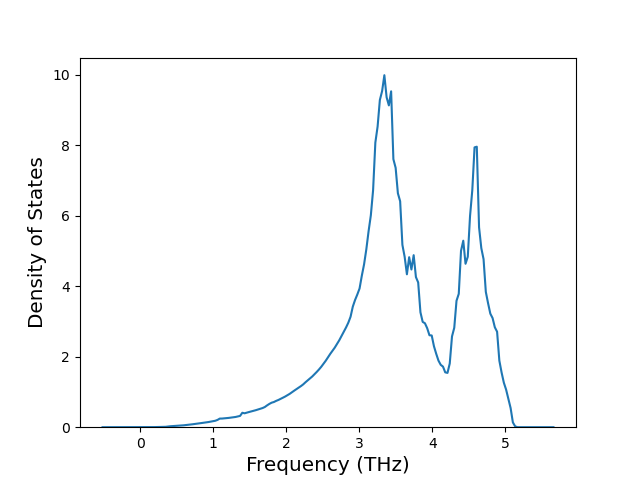
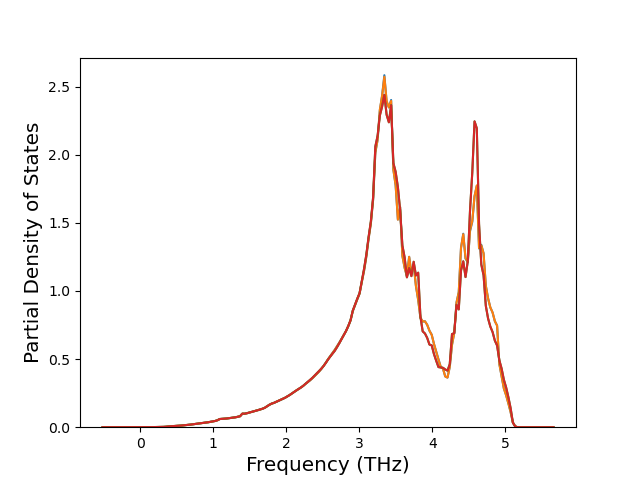
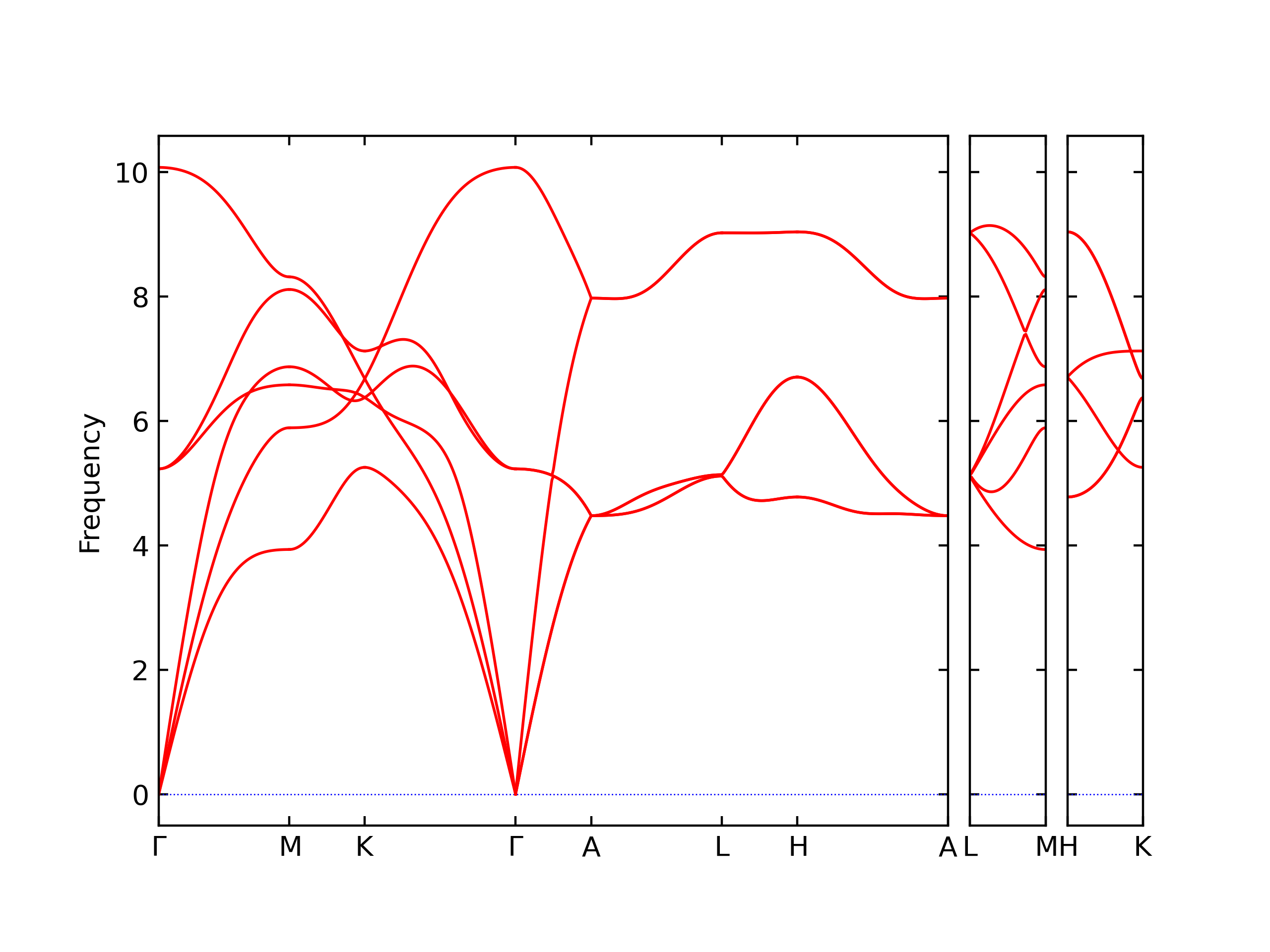
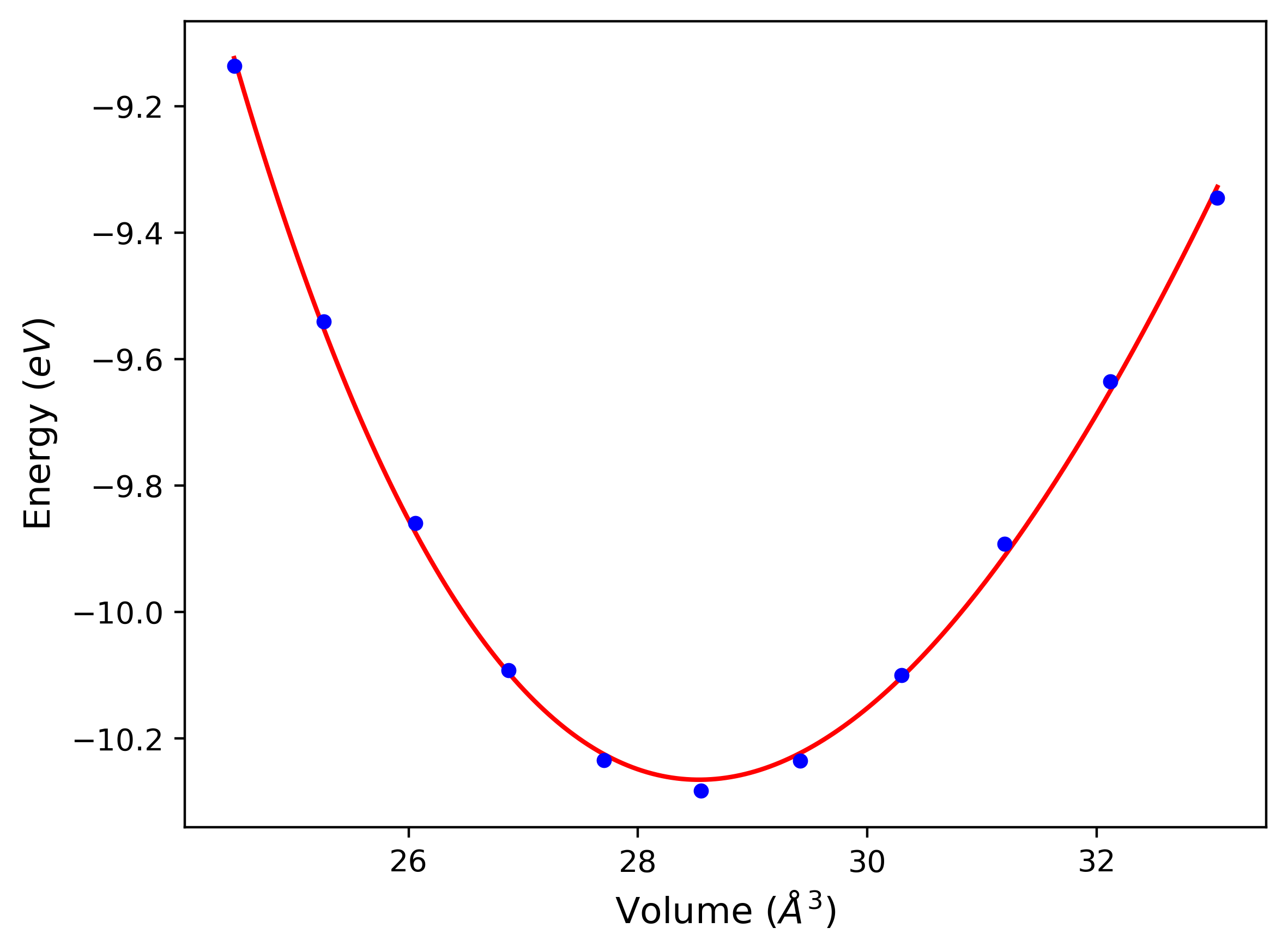
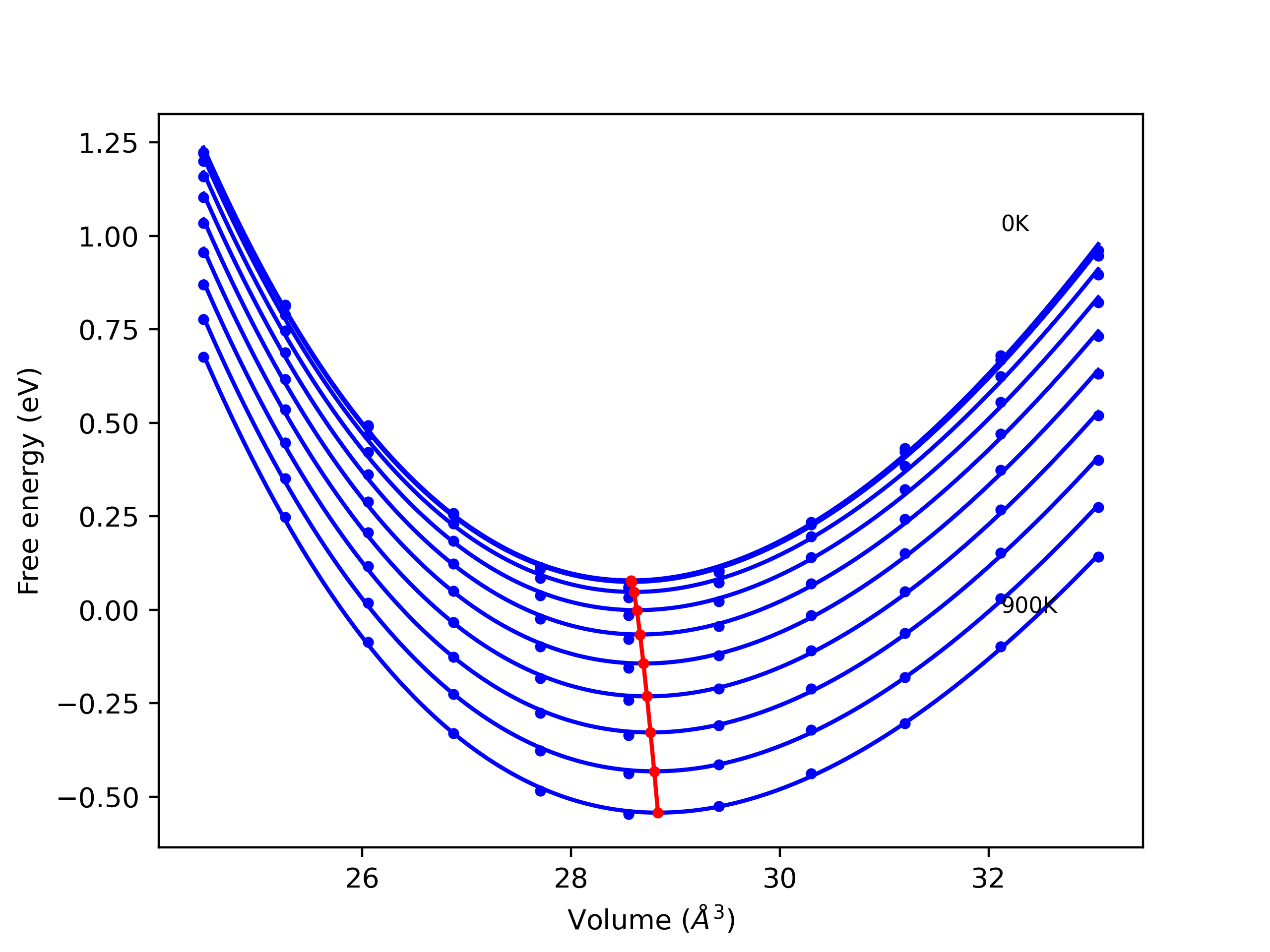
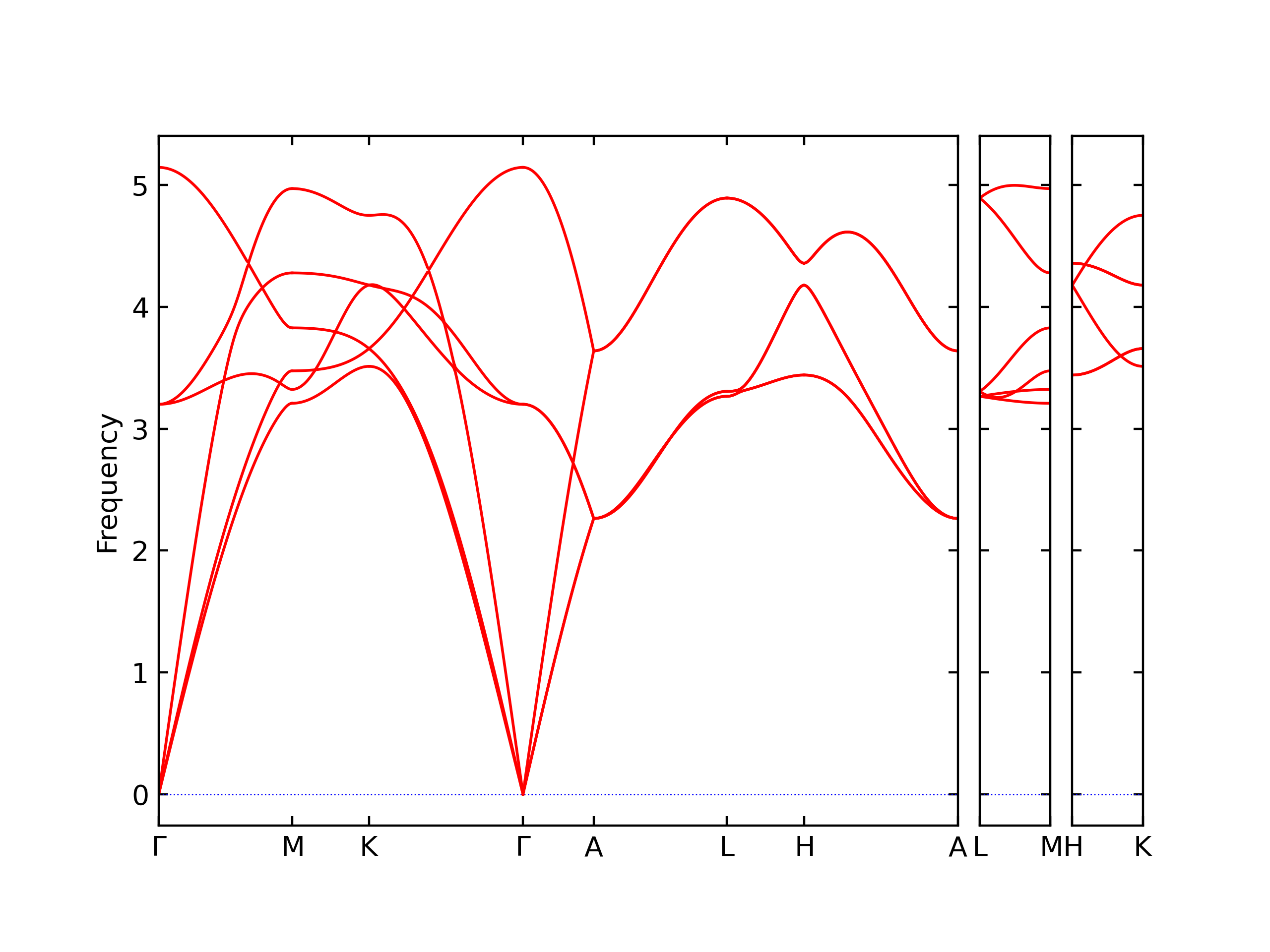
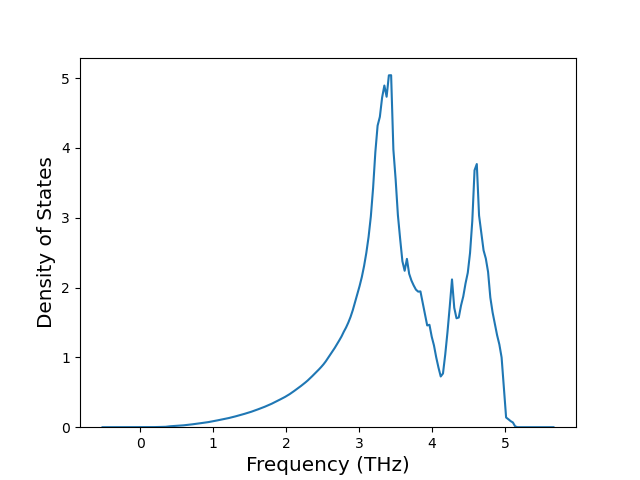
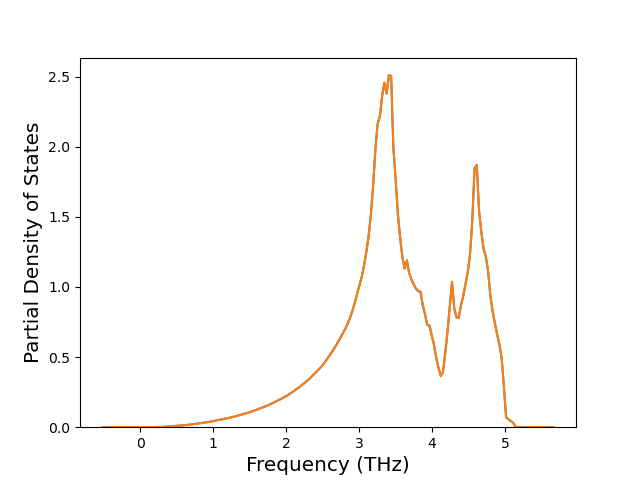
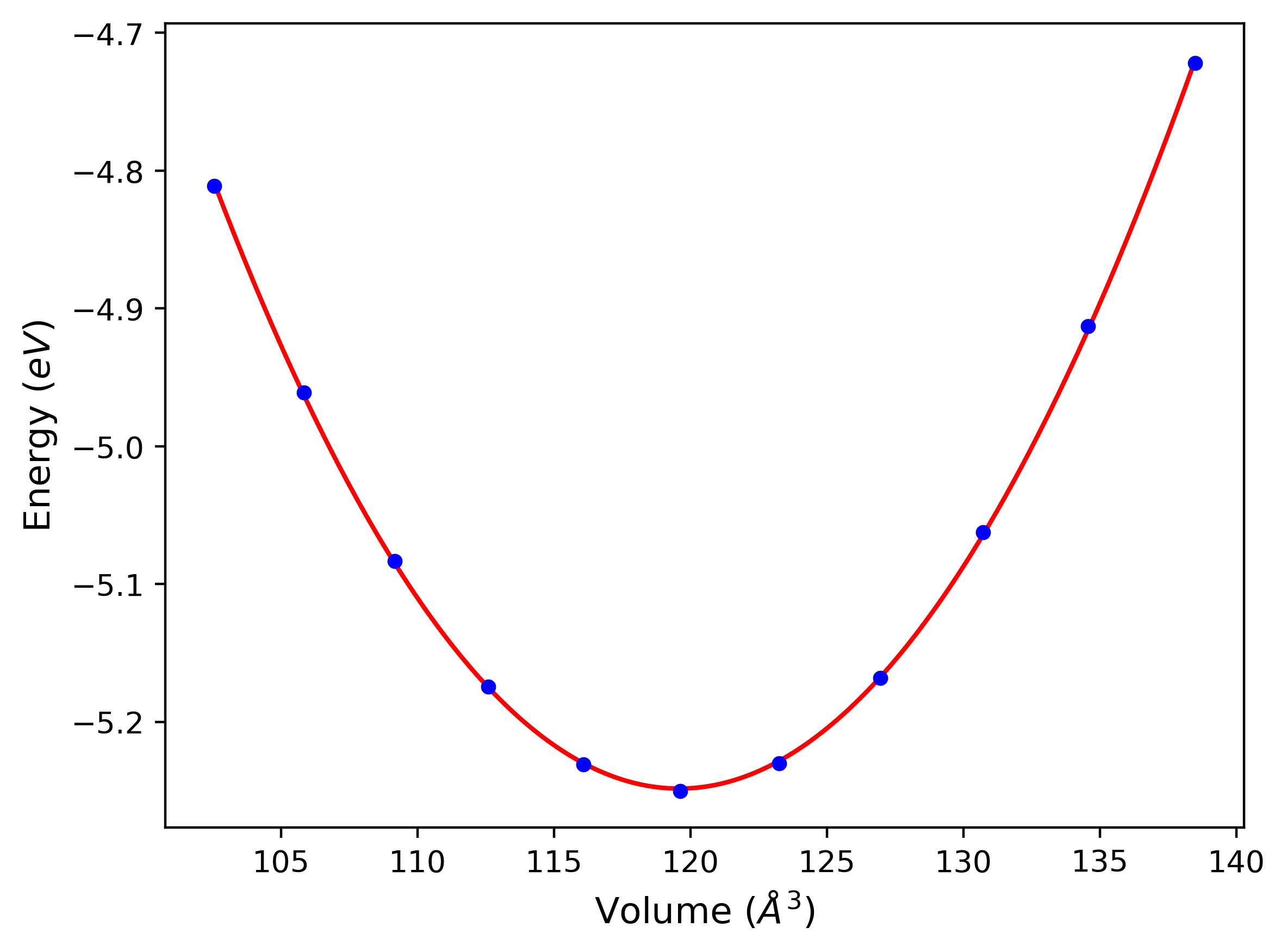
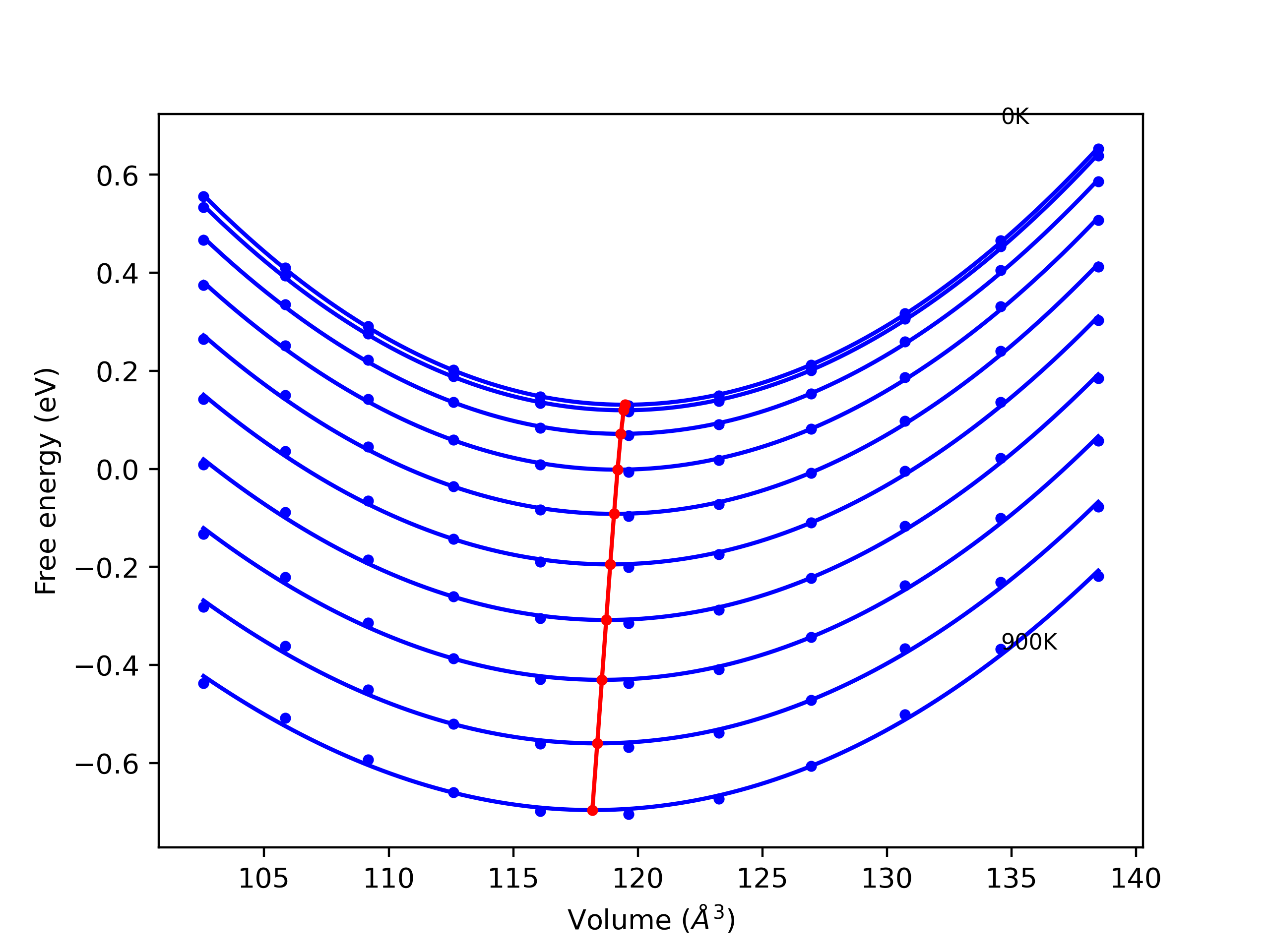
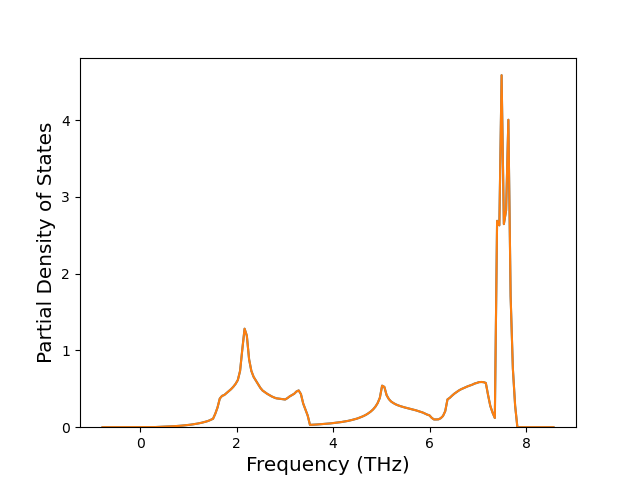
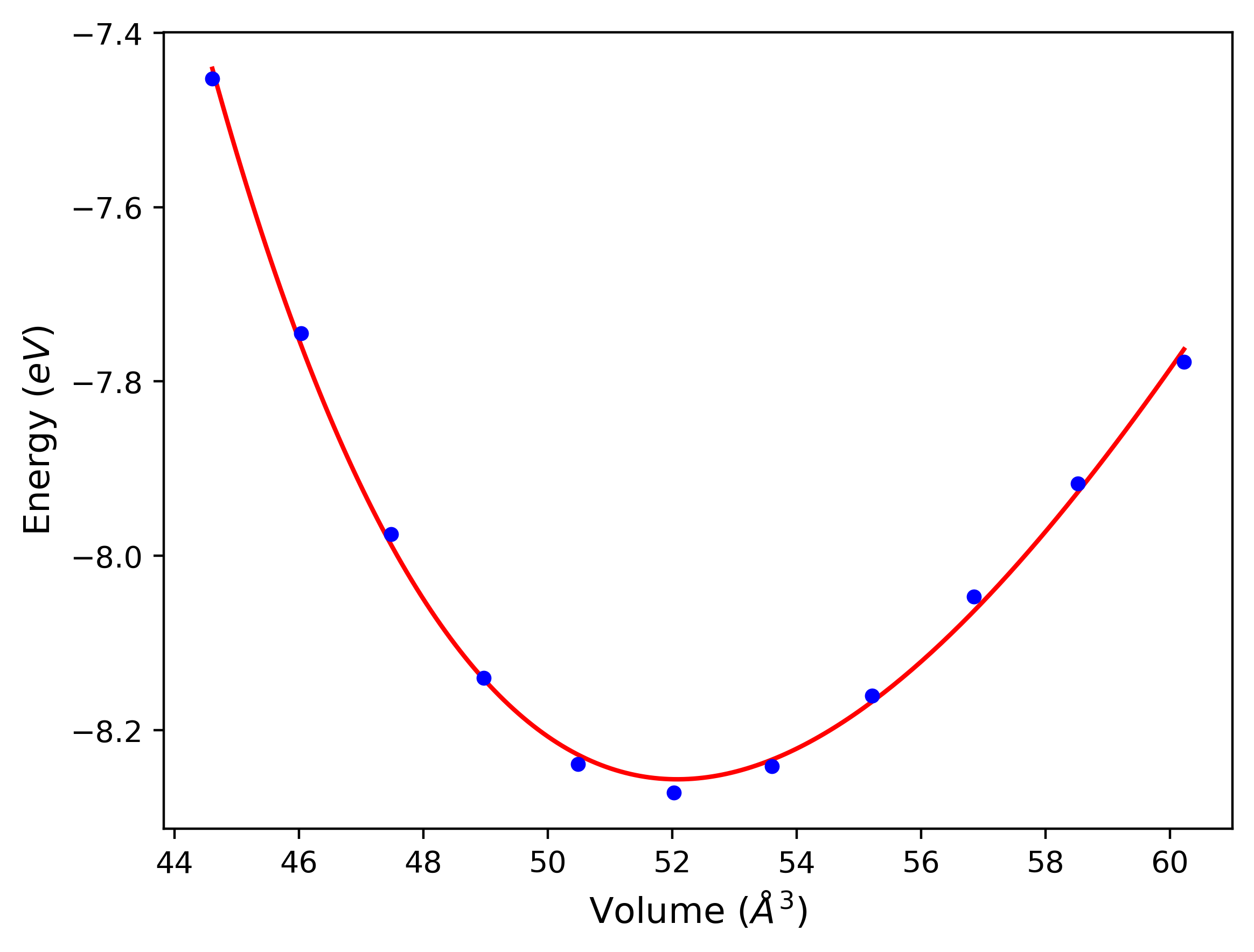
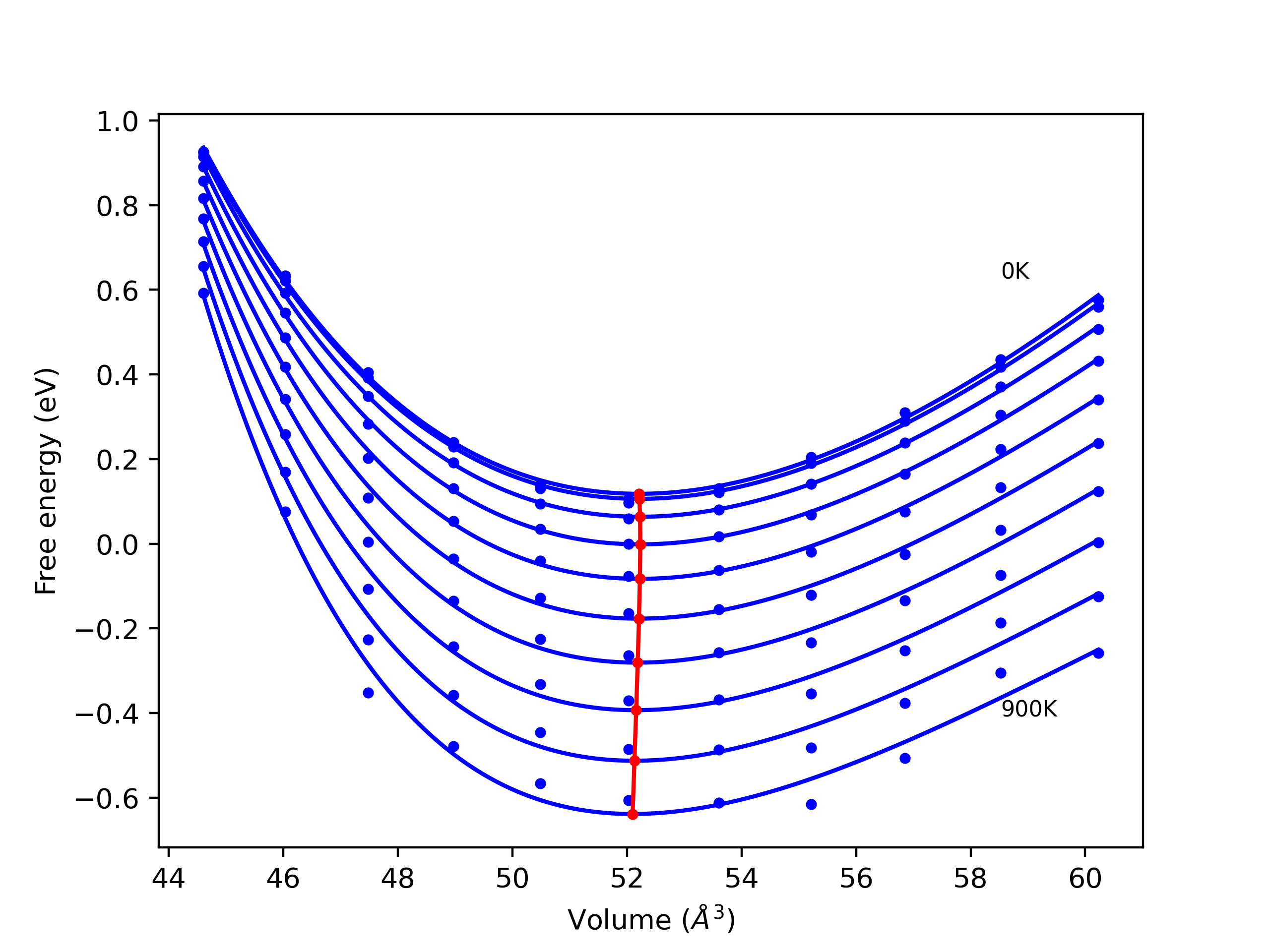
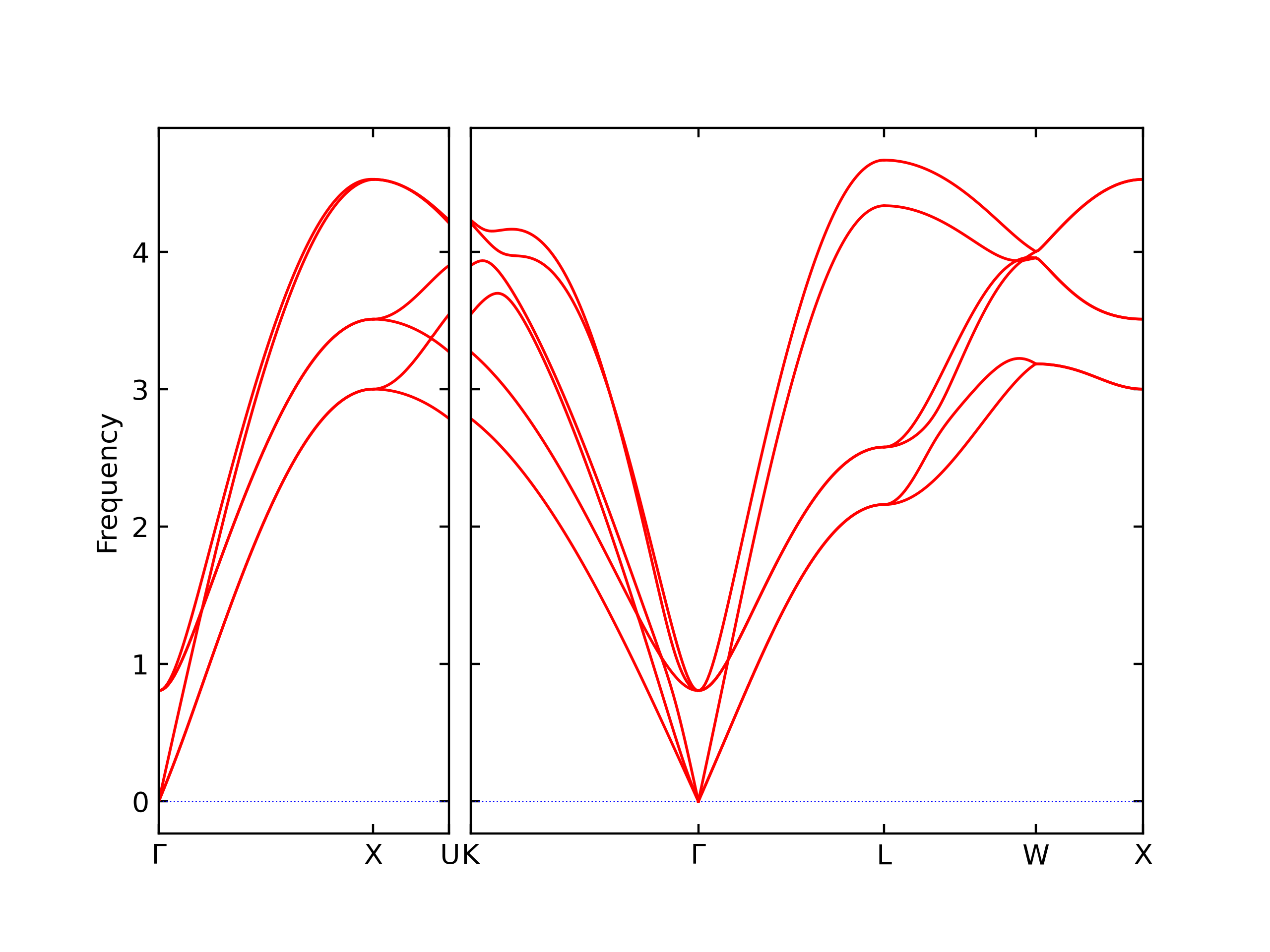
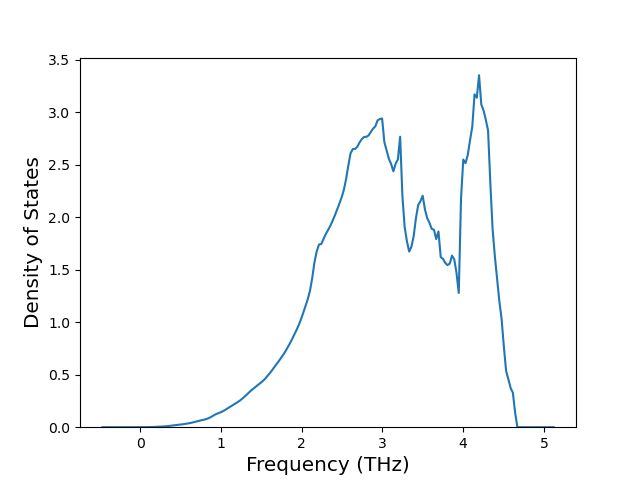
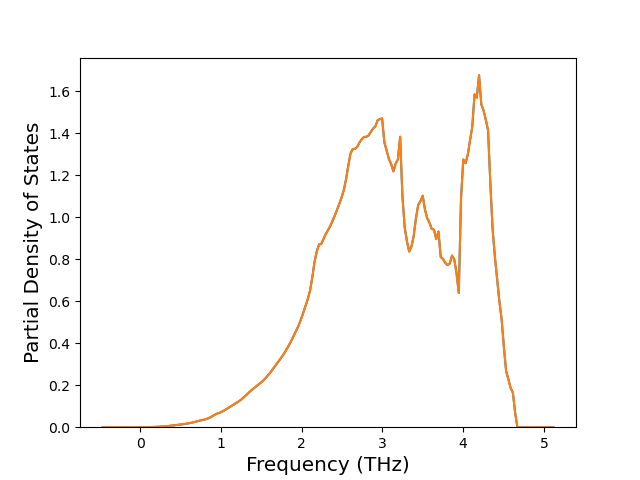
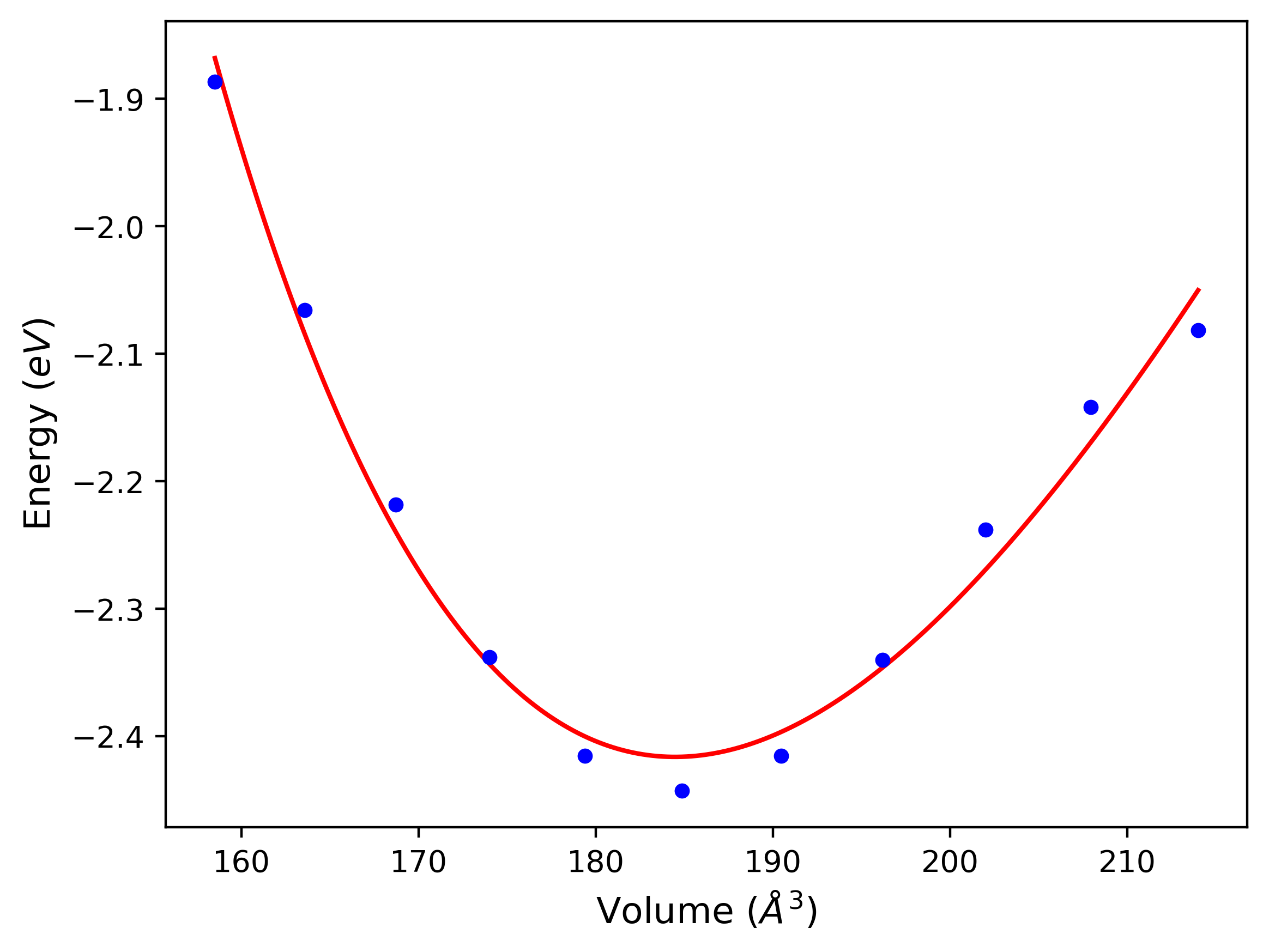
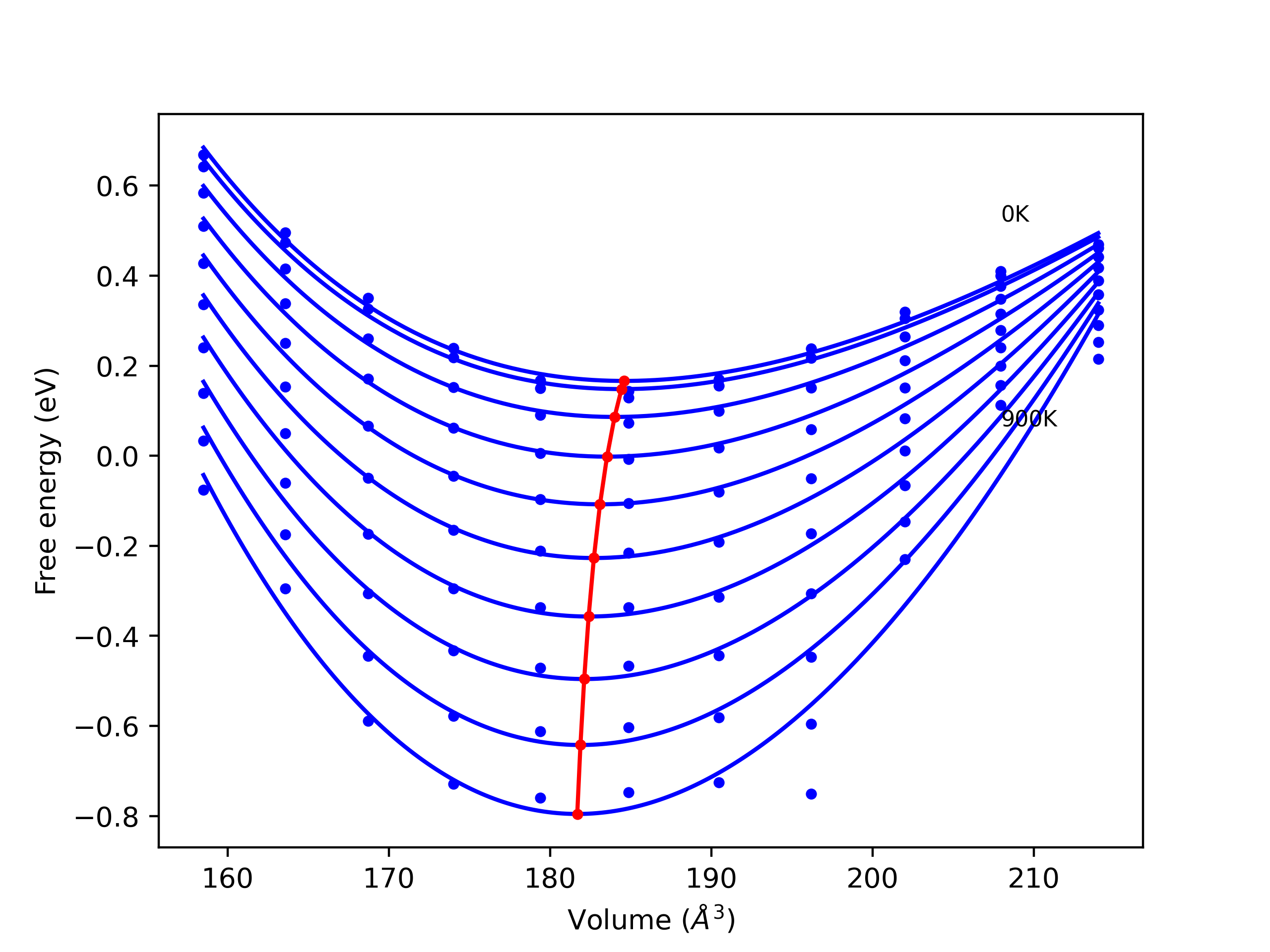
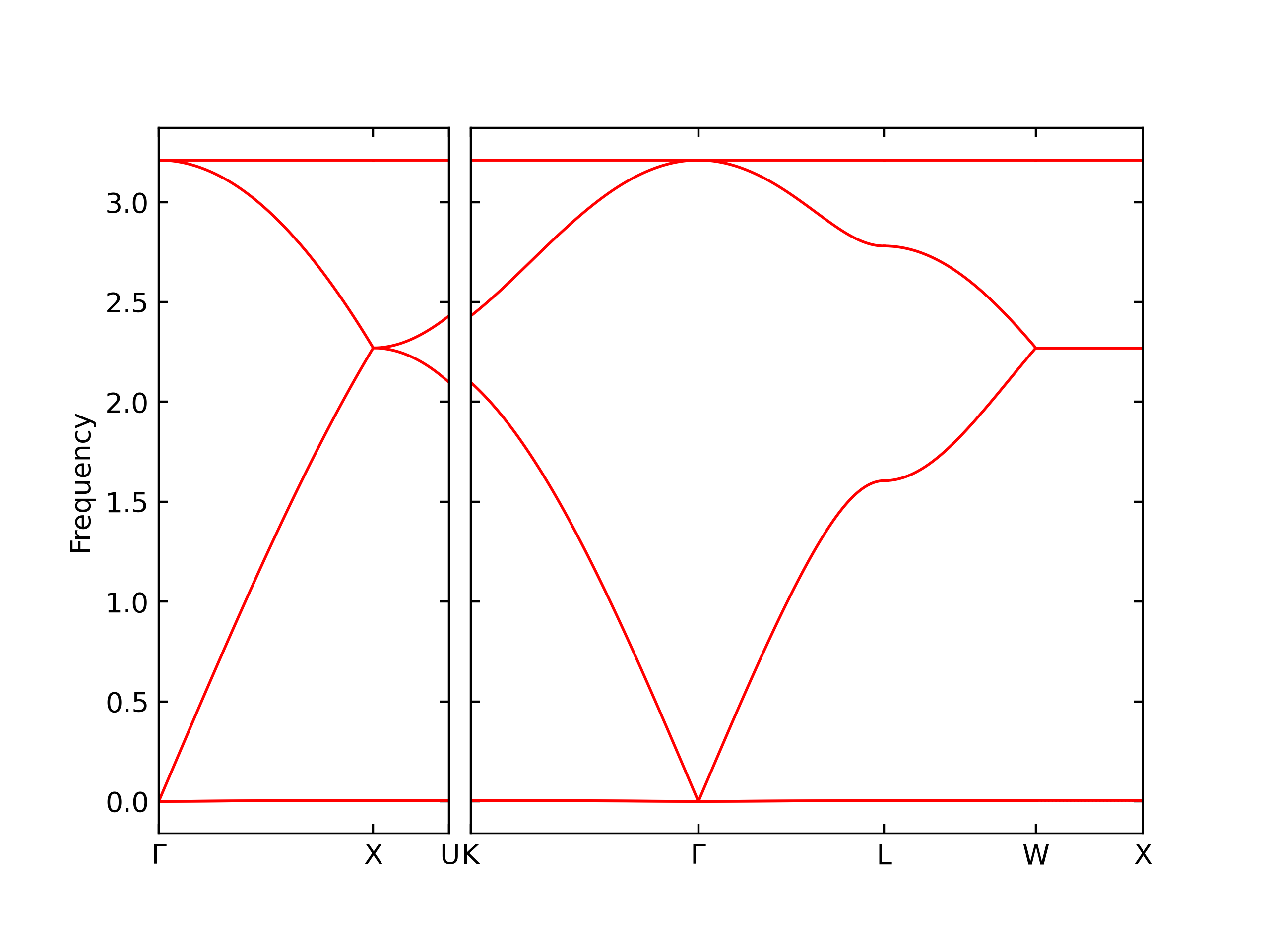
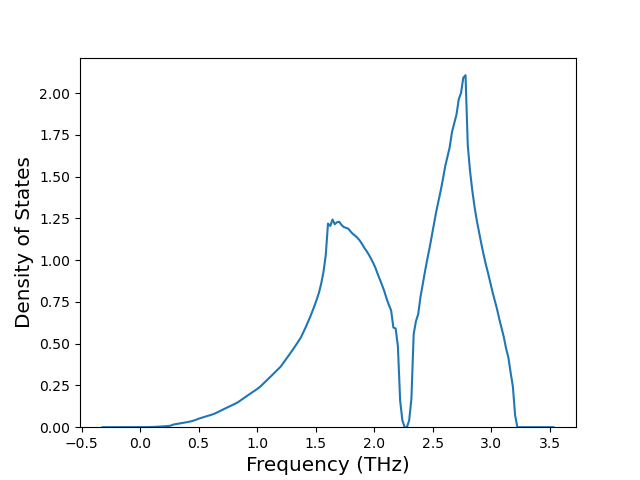
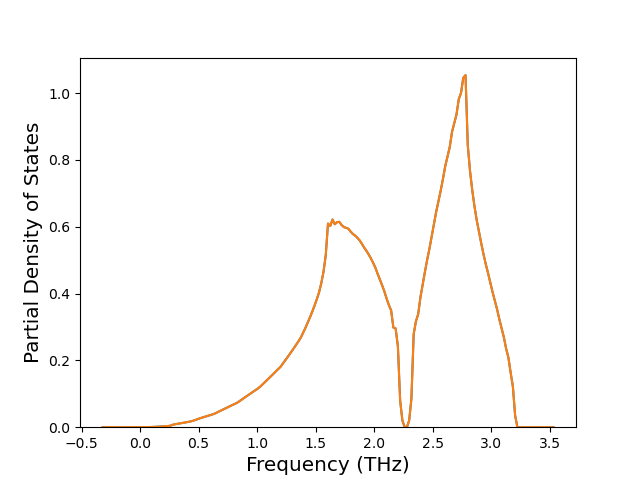
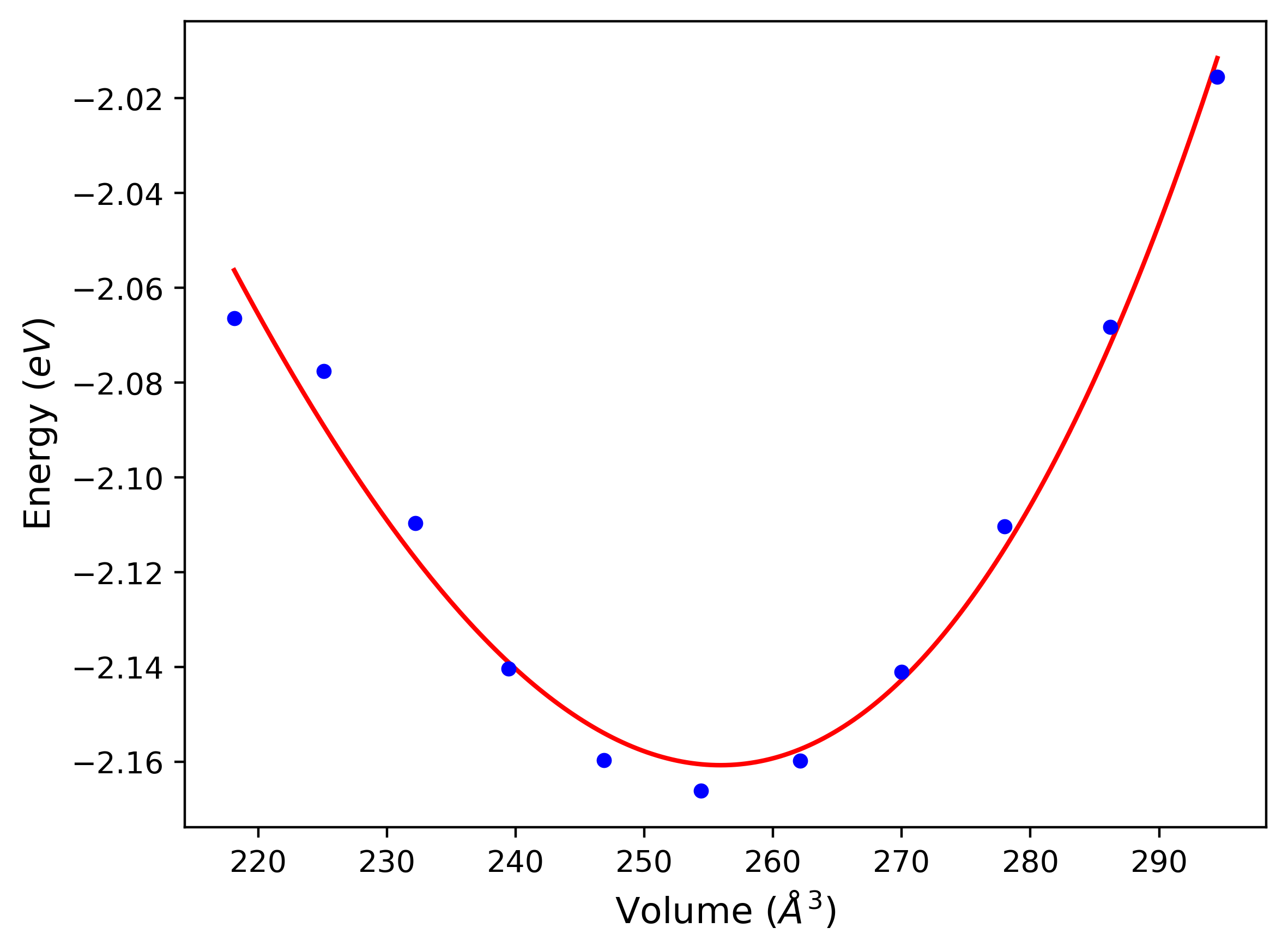
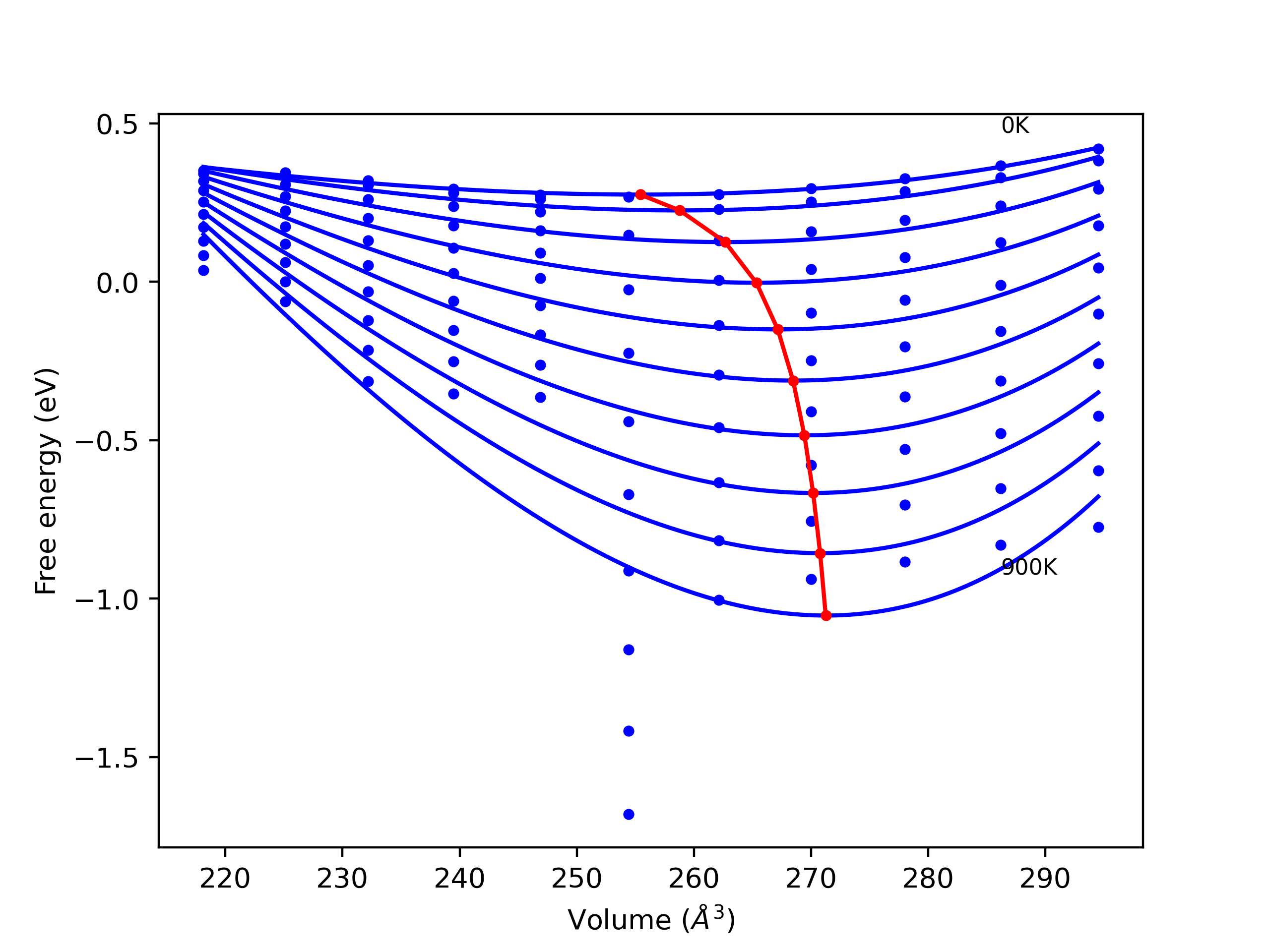
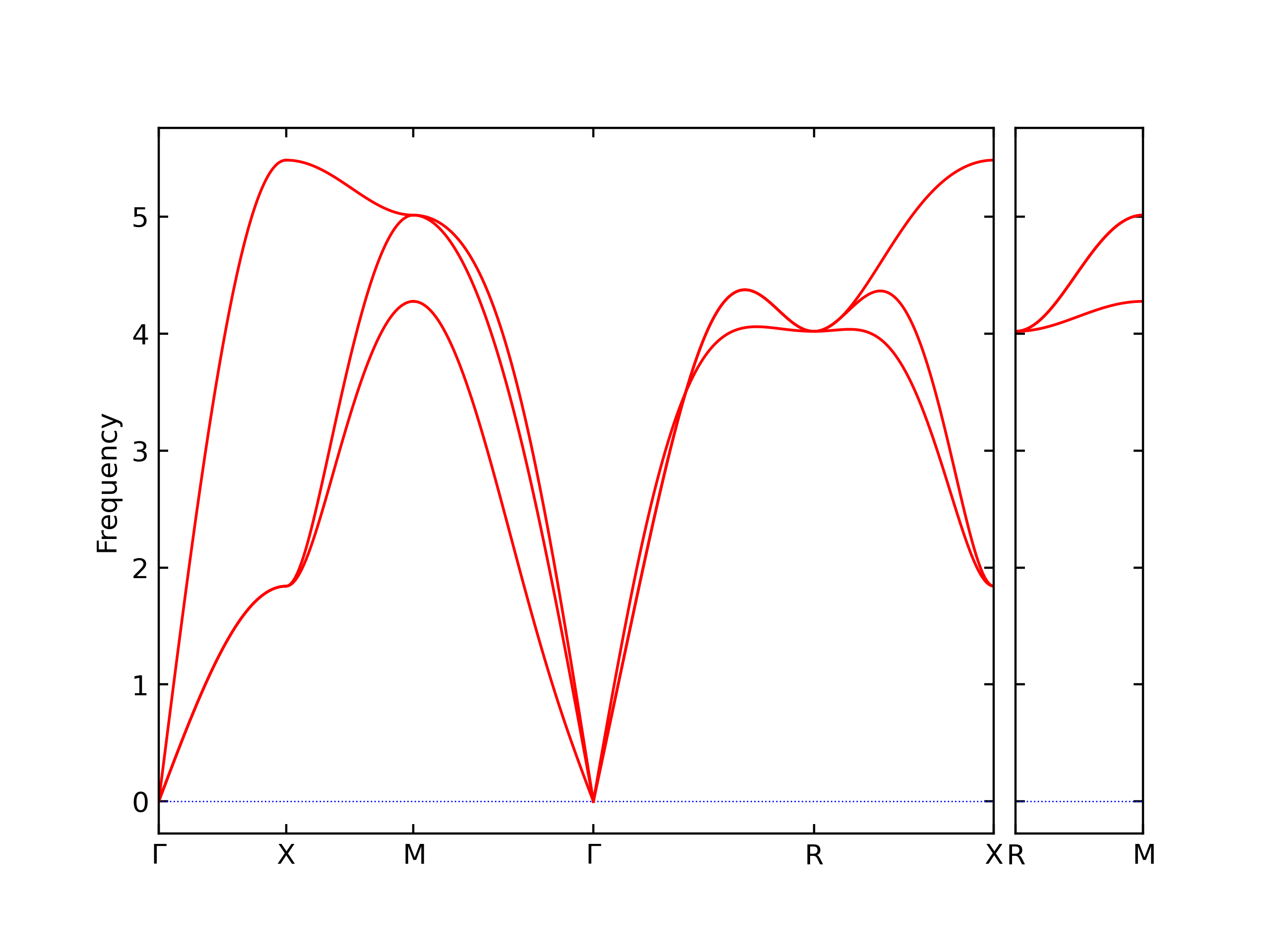
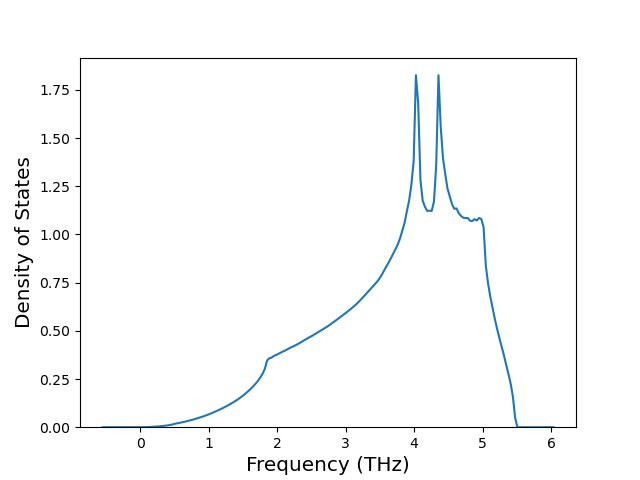
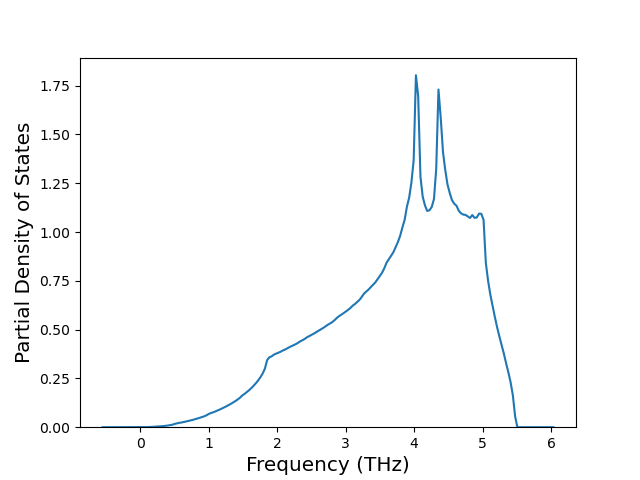
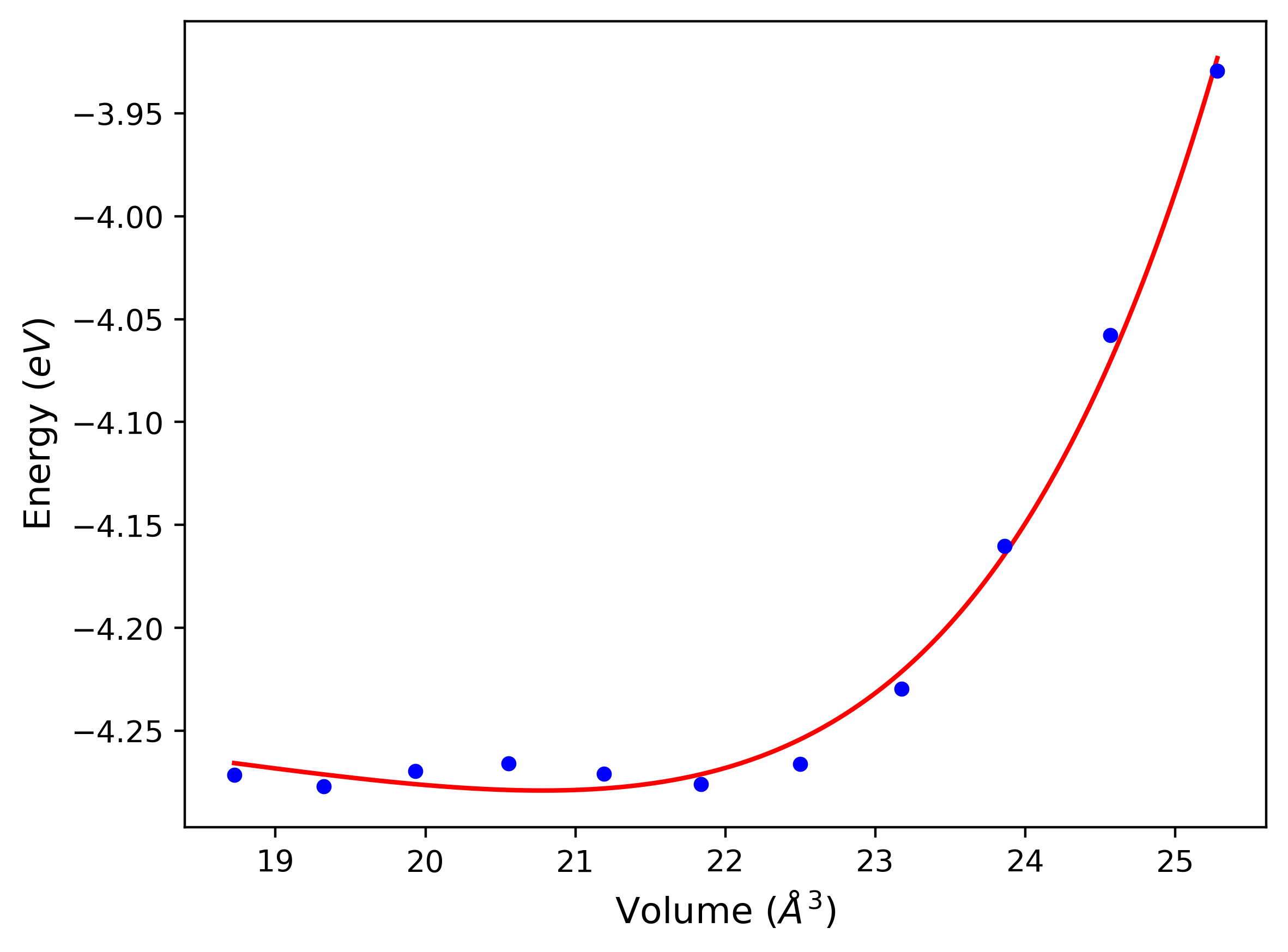
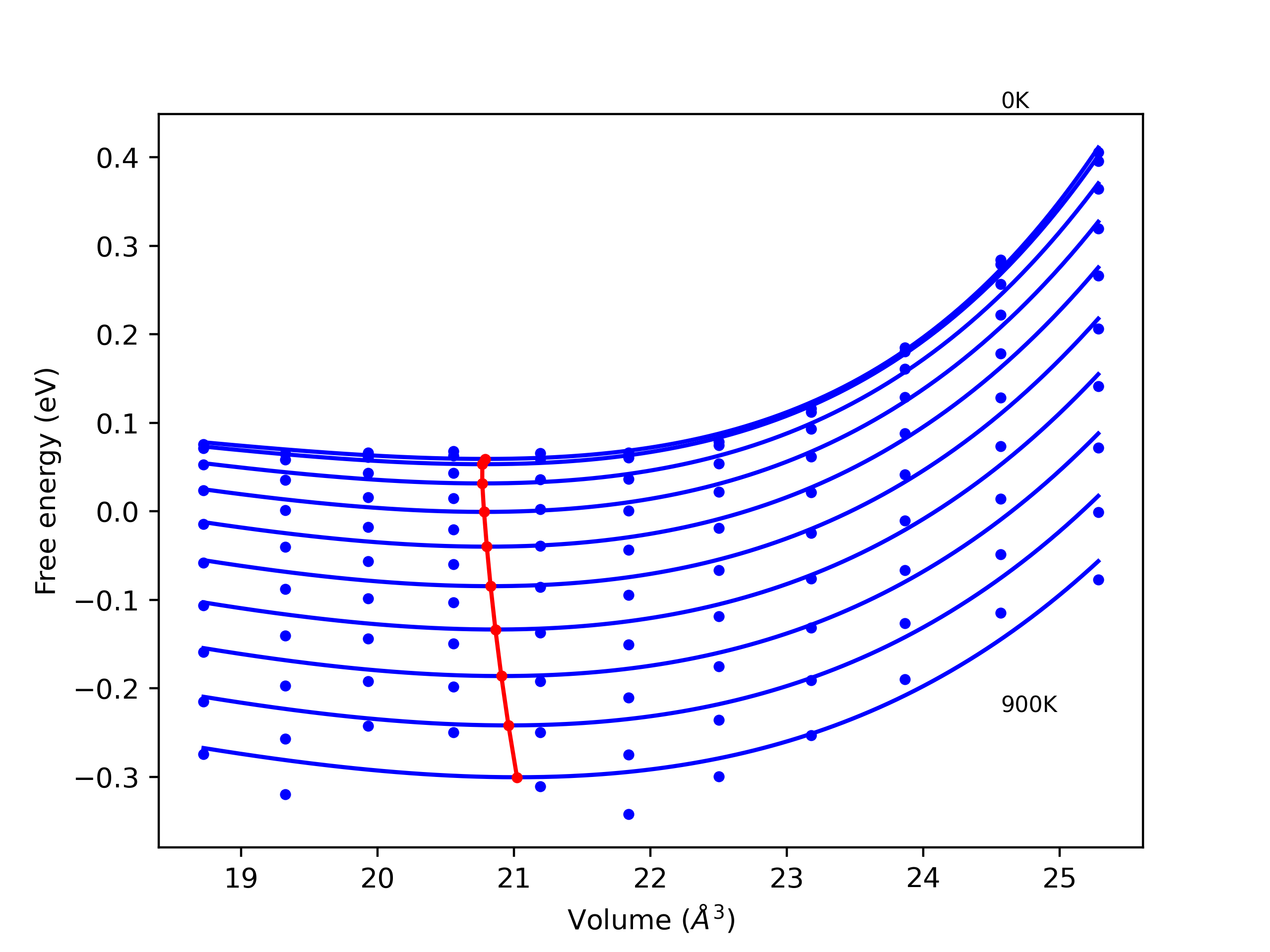
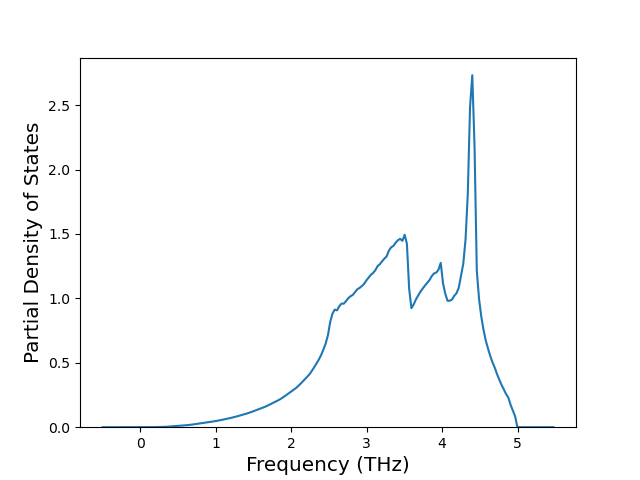
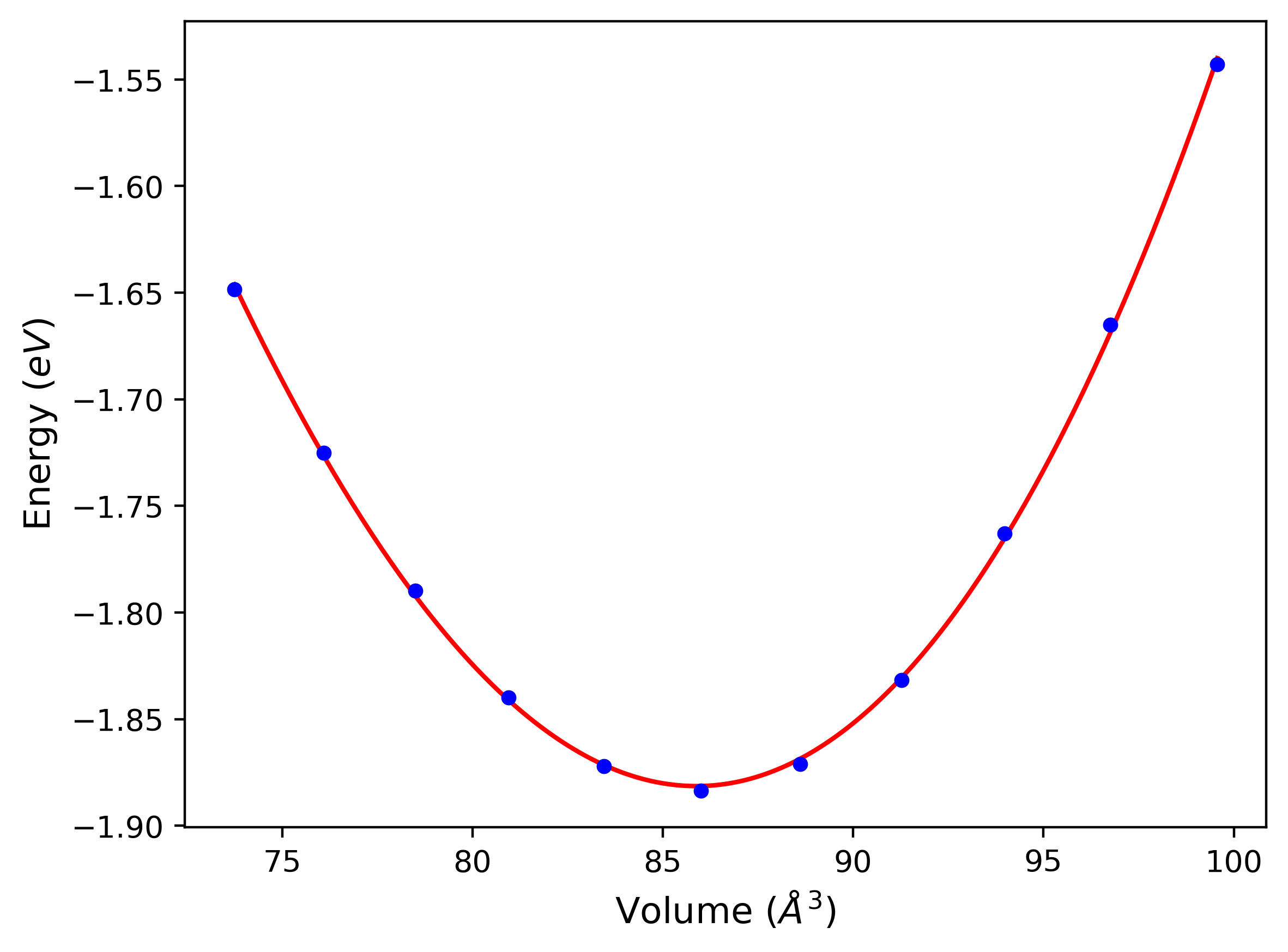
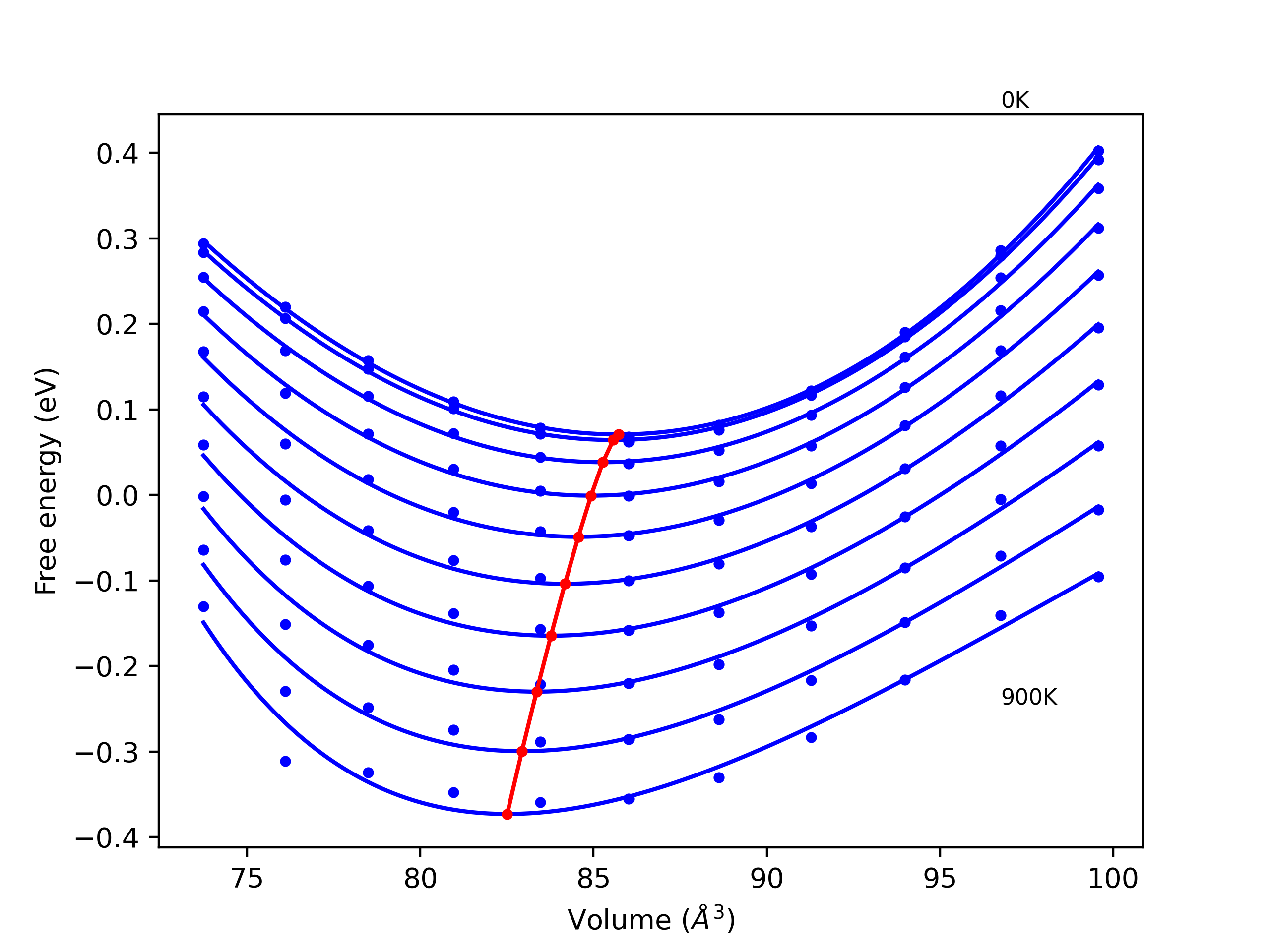
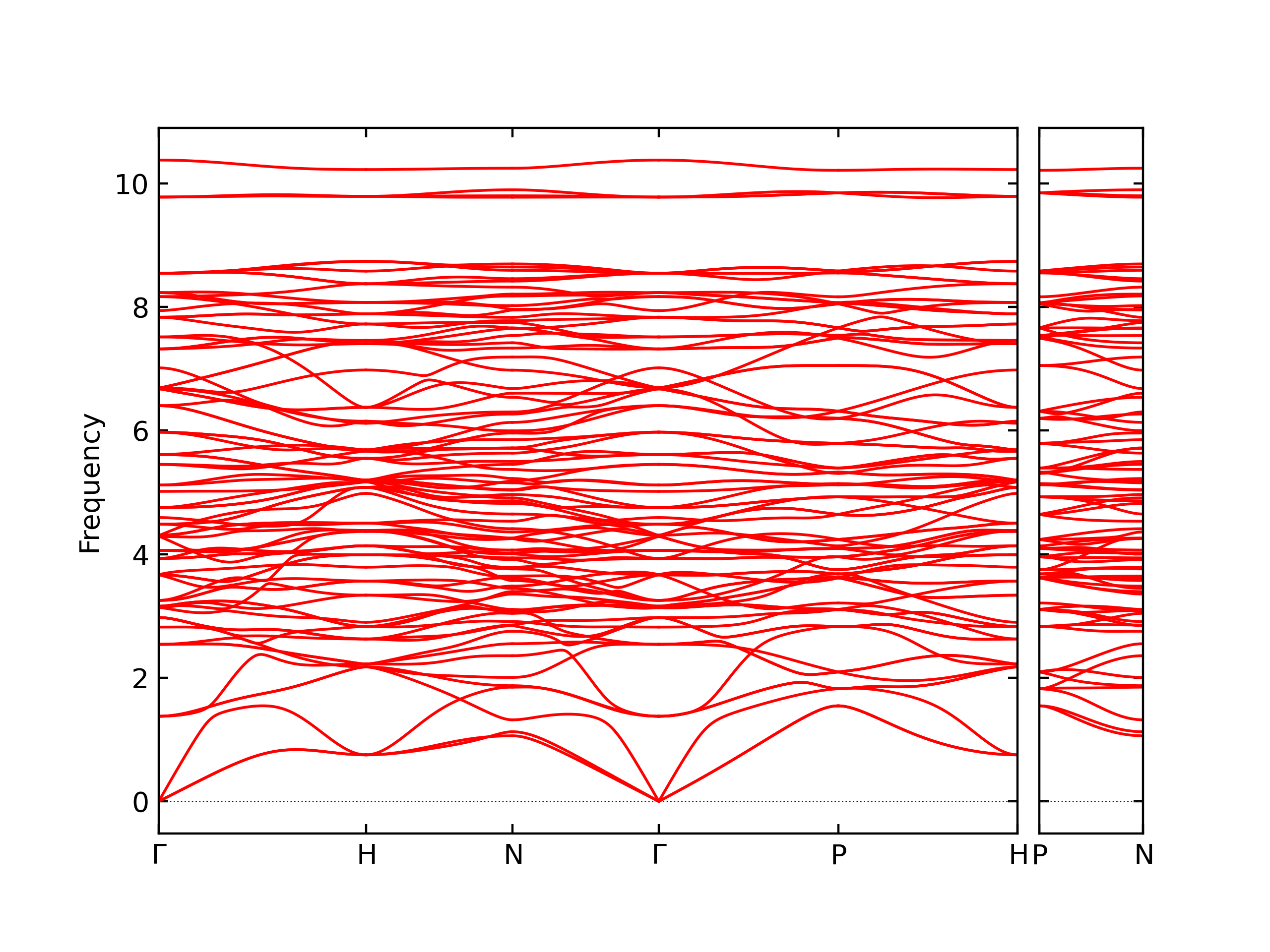
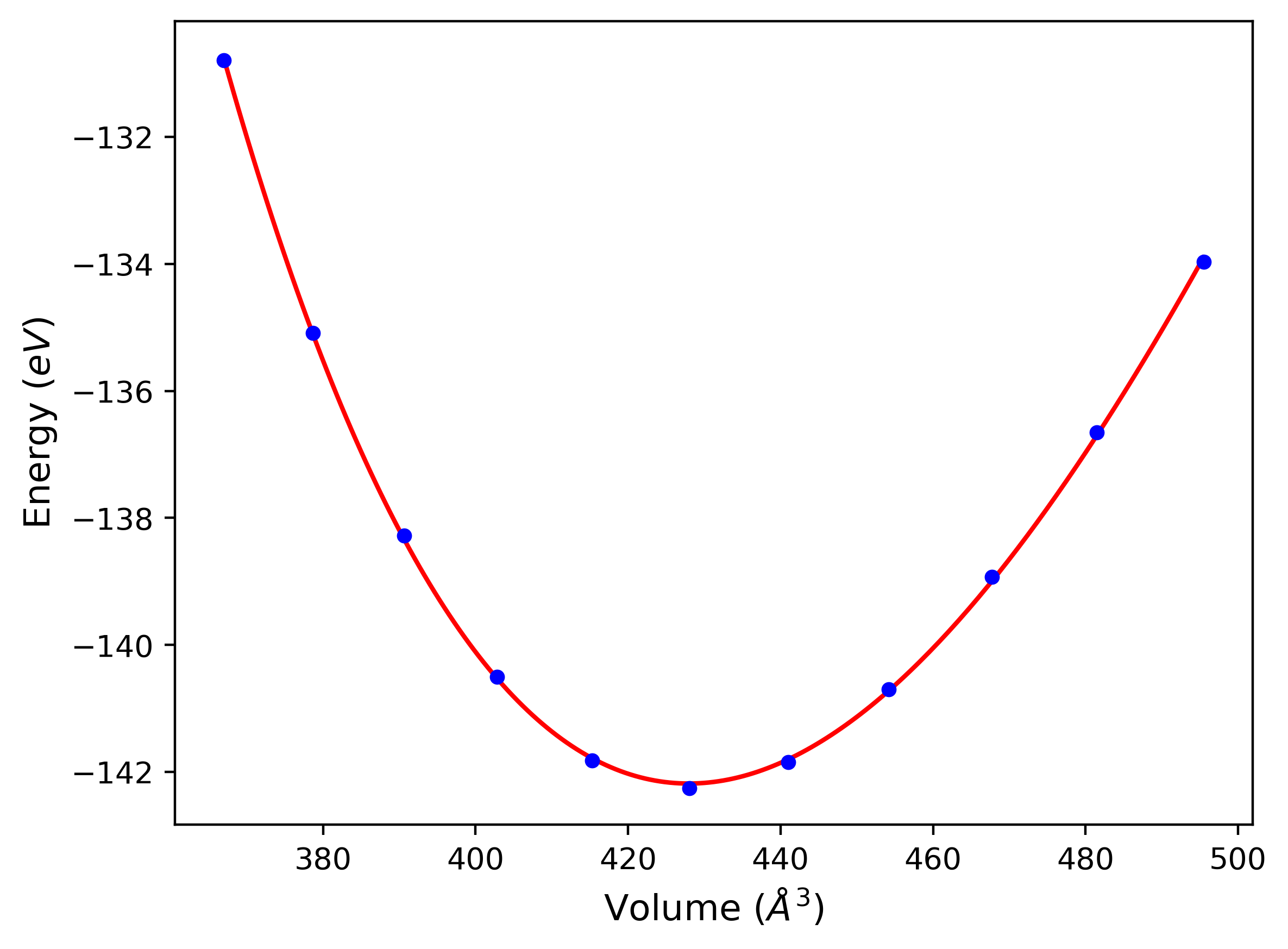
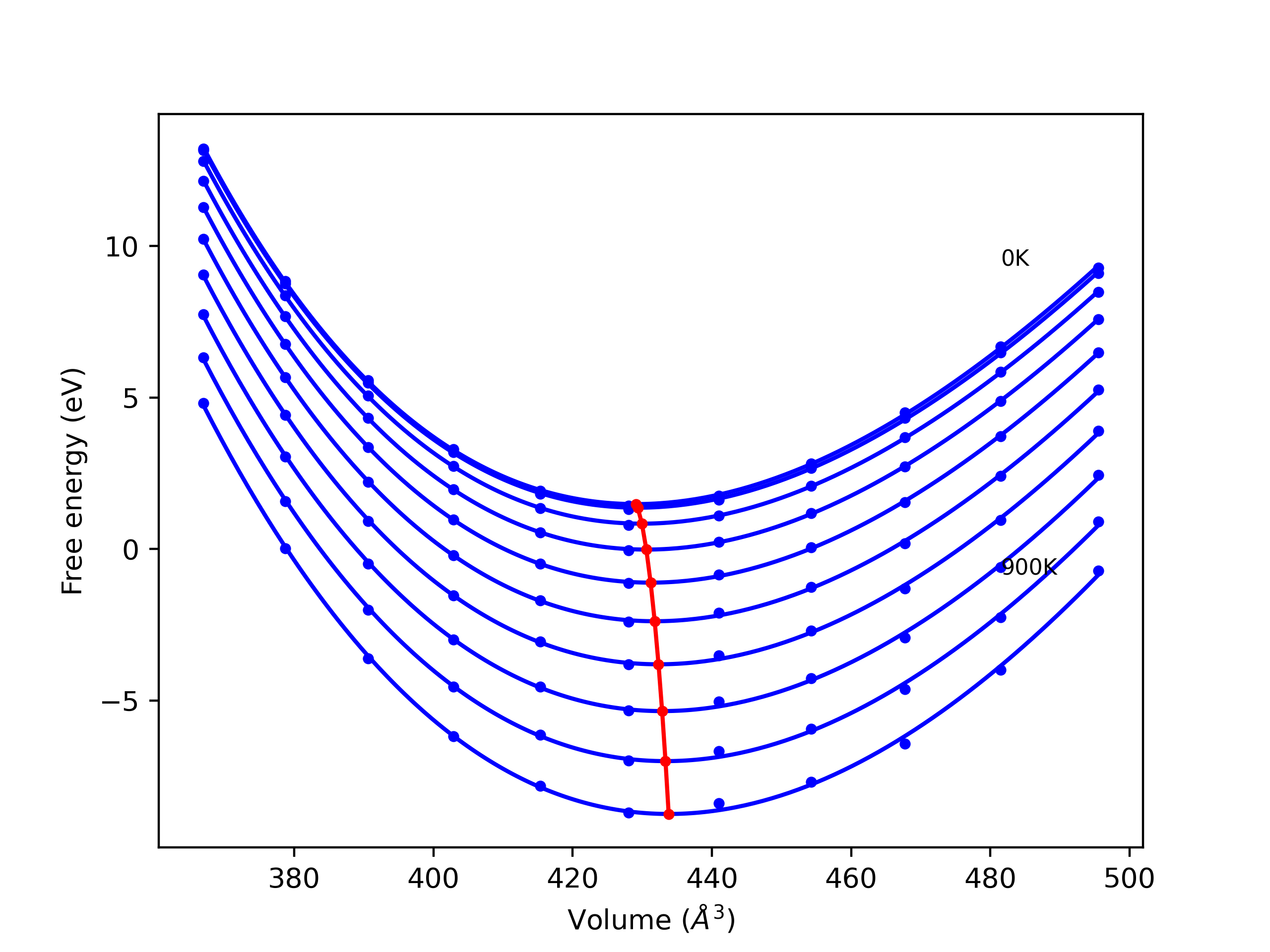
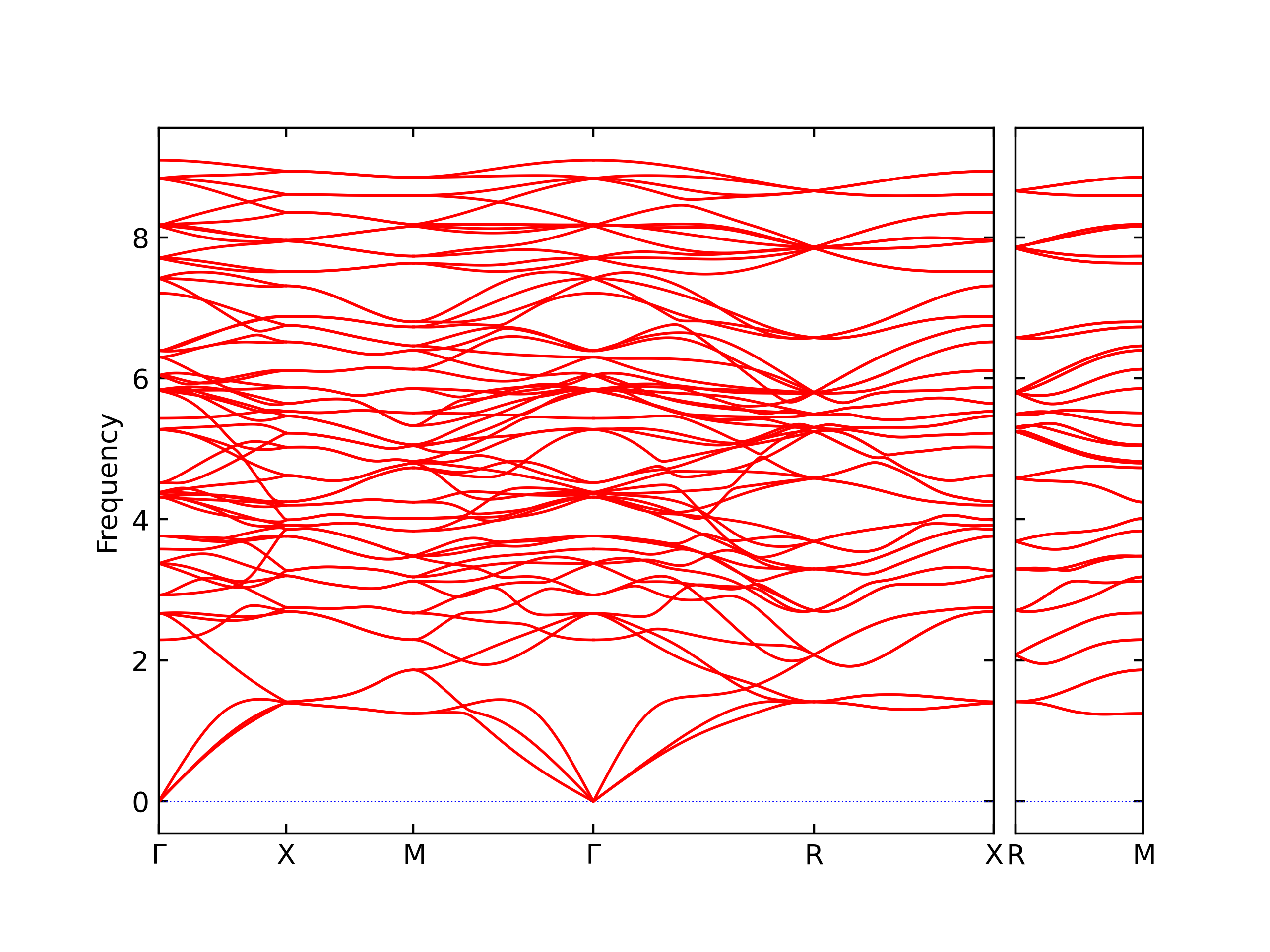
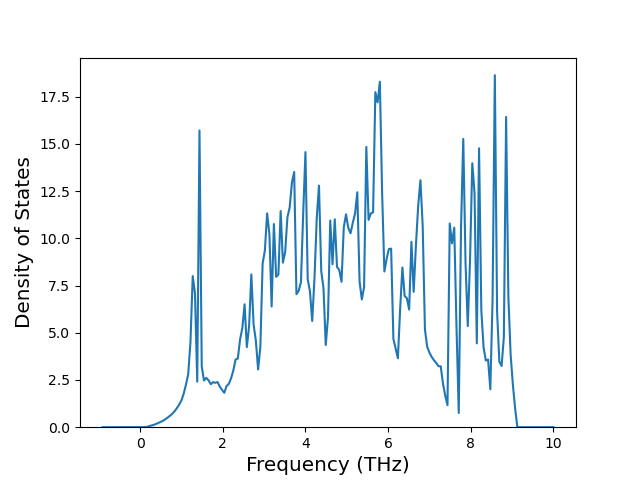
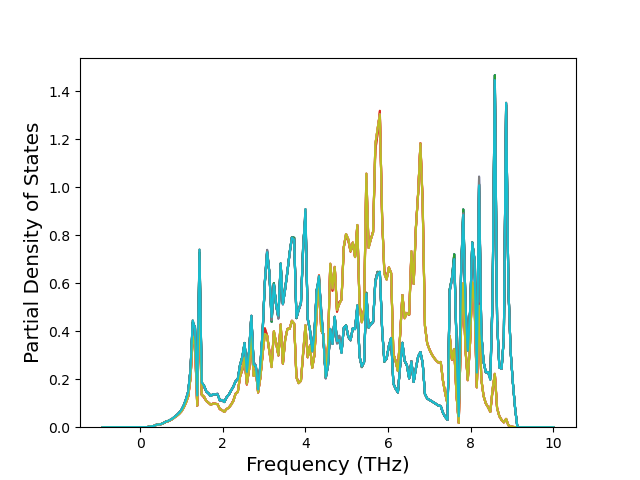
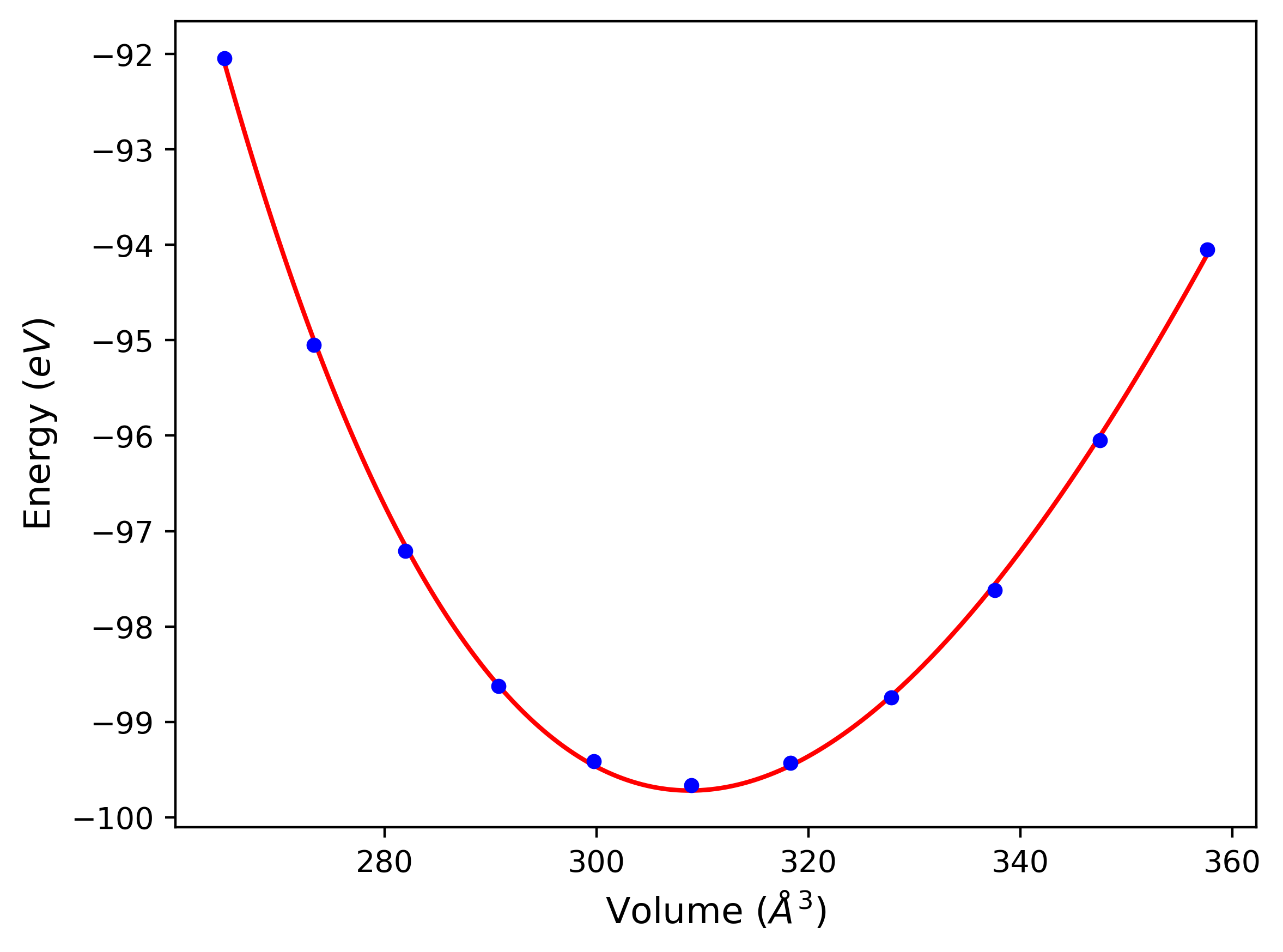
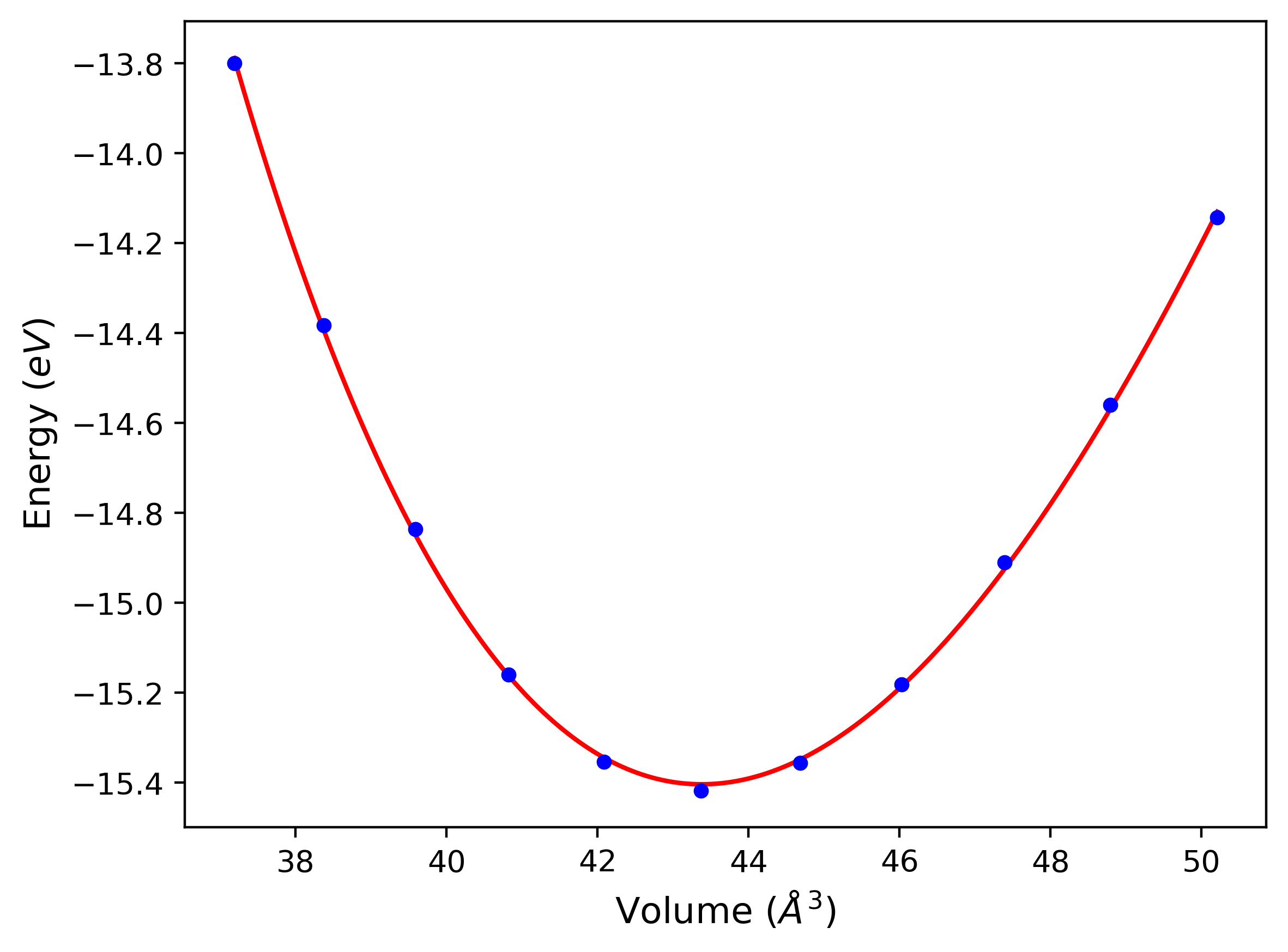
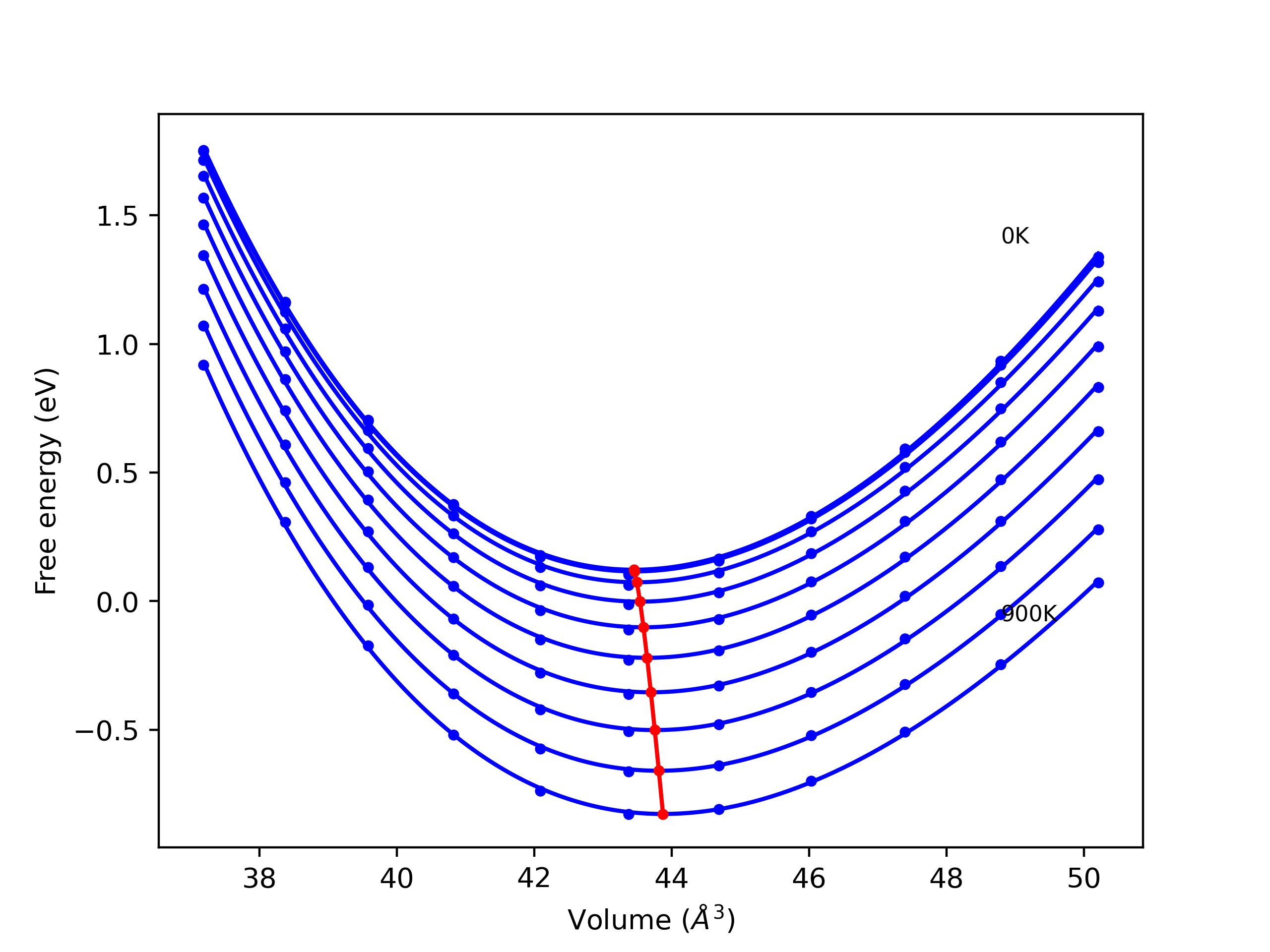
Thermodynamic Predictions
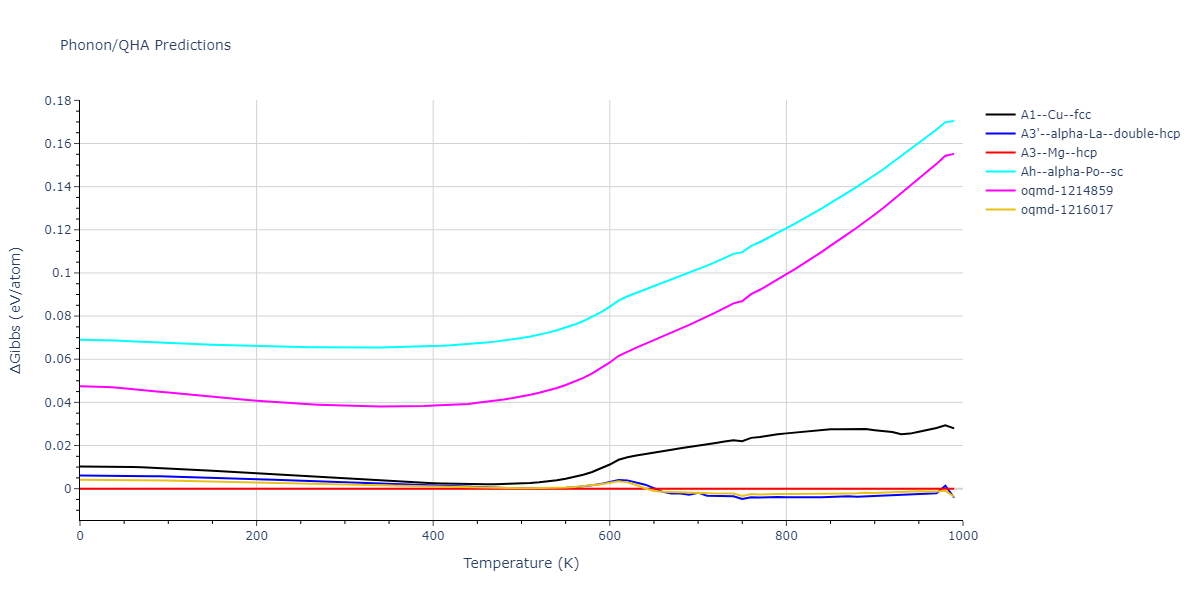
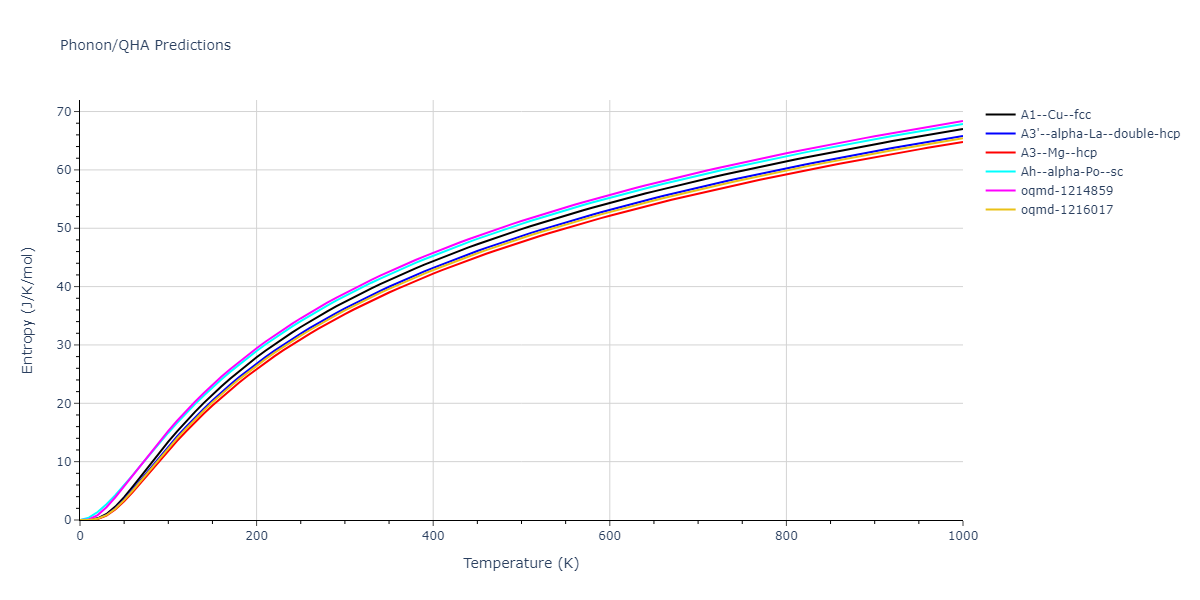
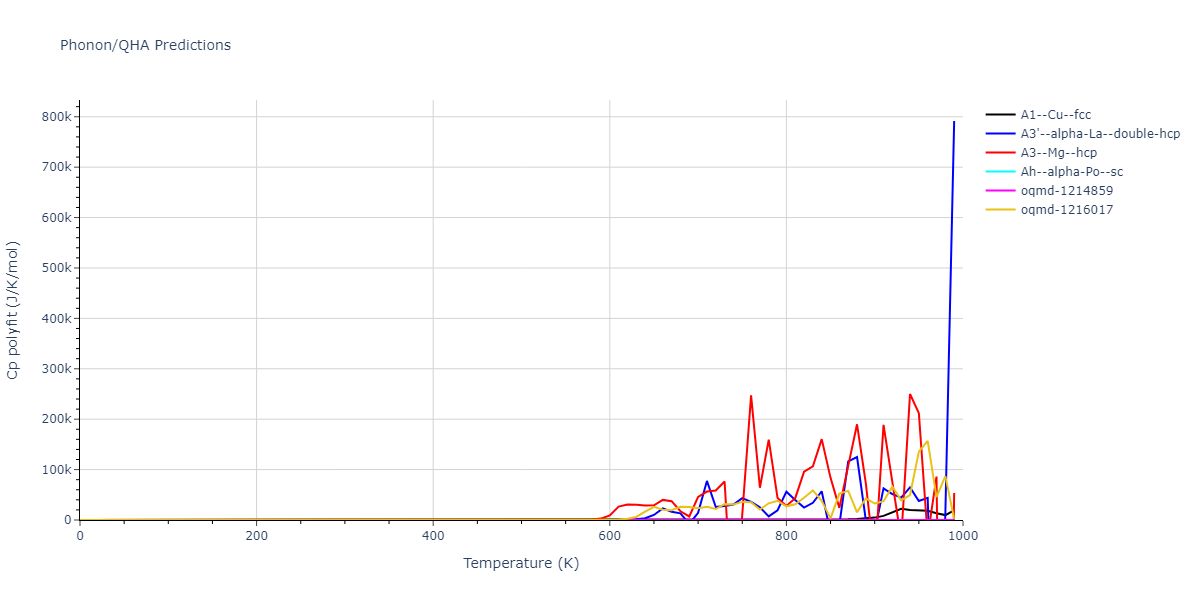
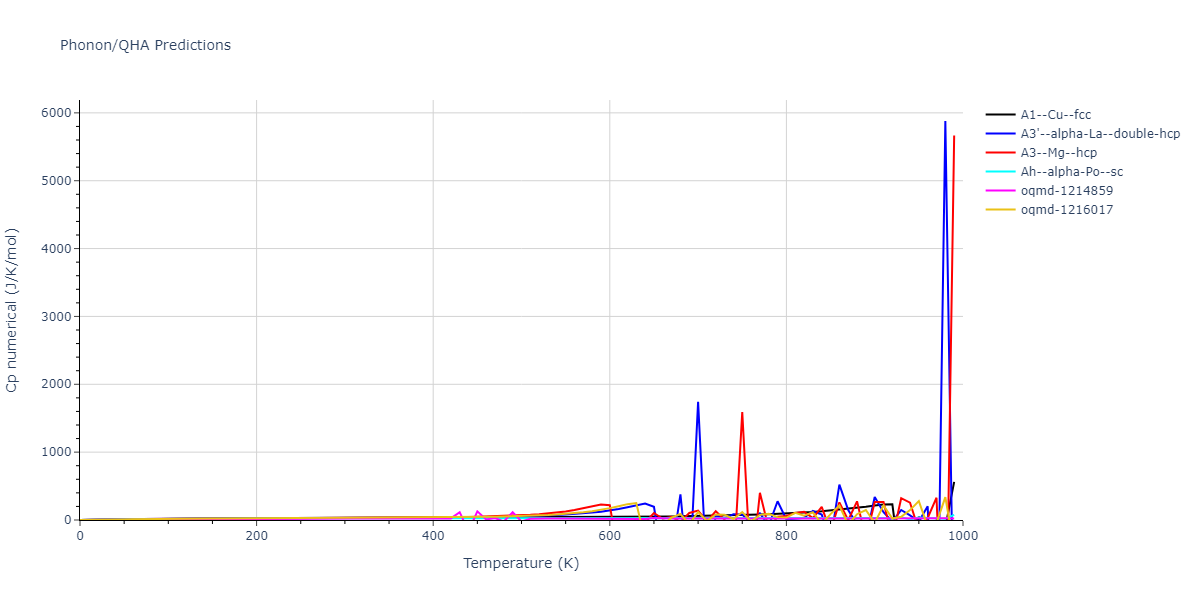
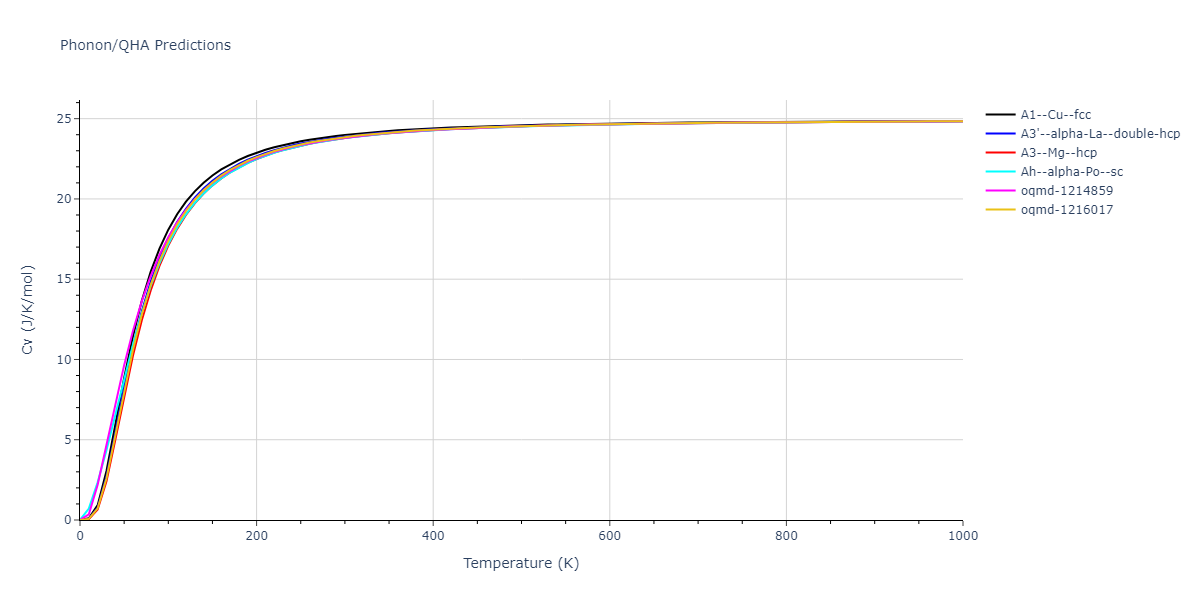
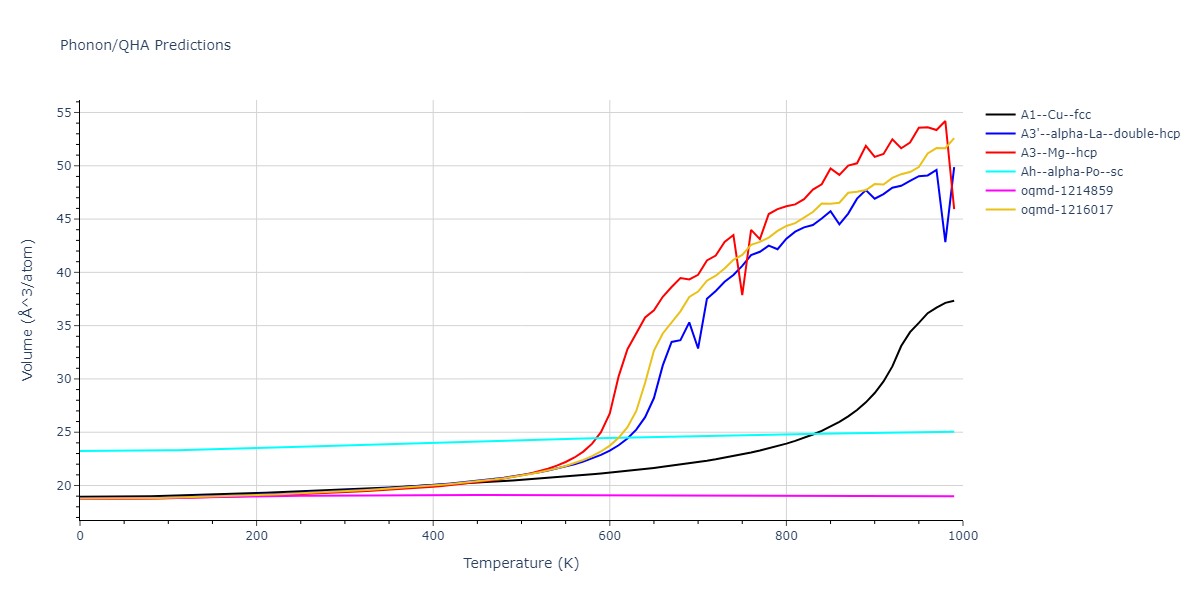
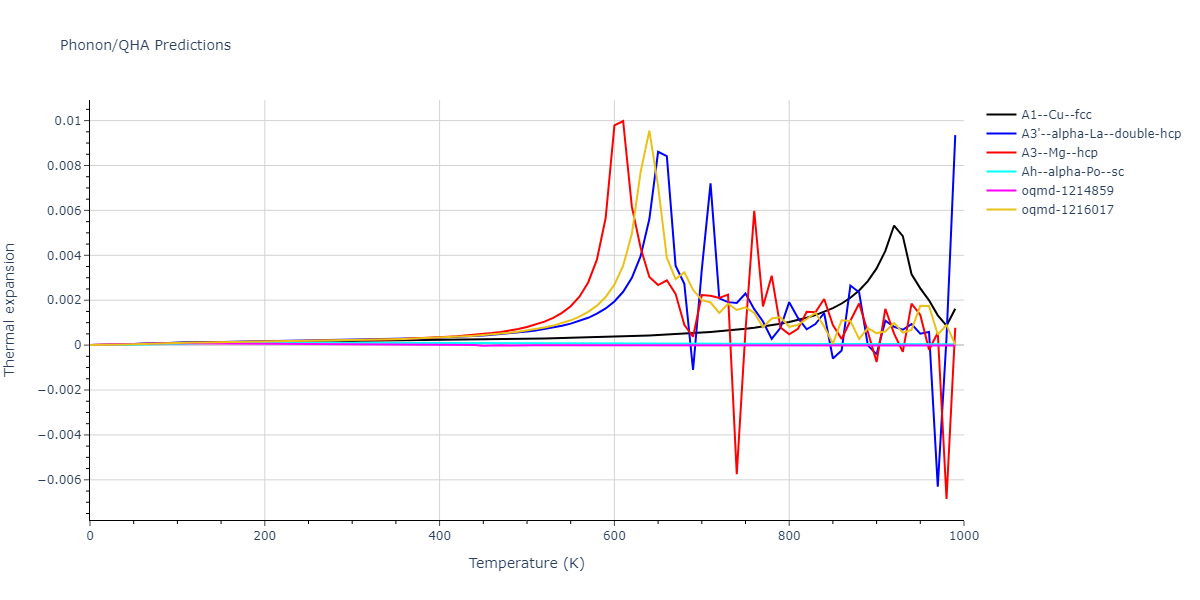
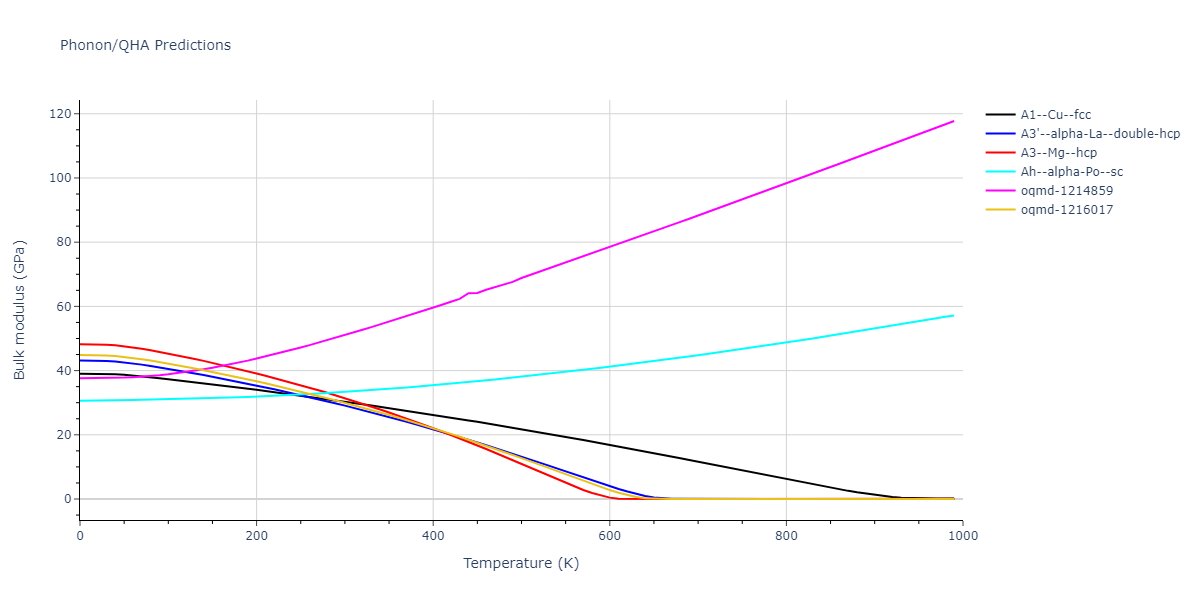
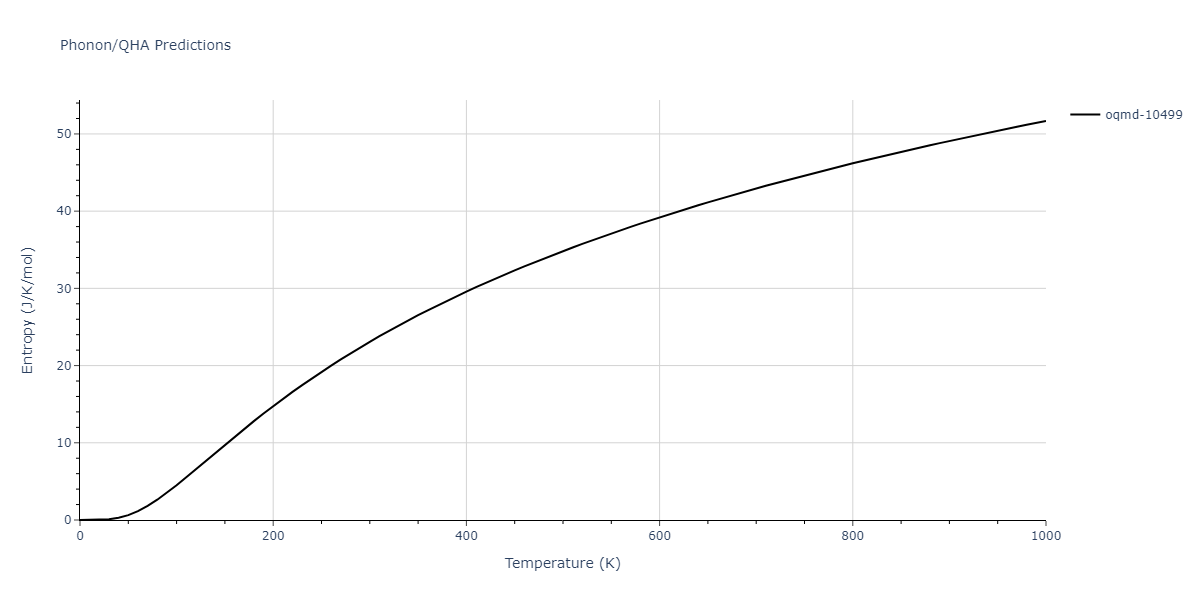
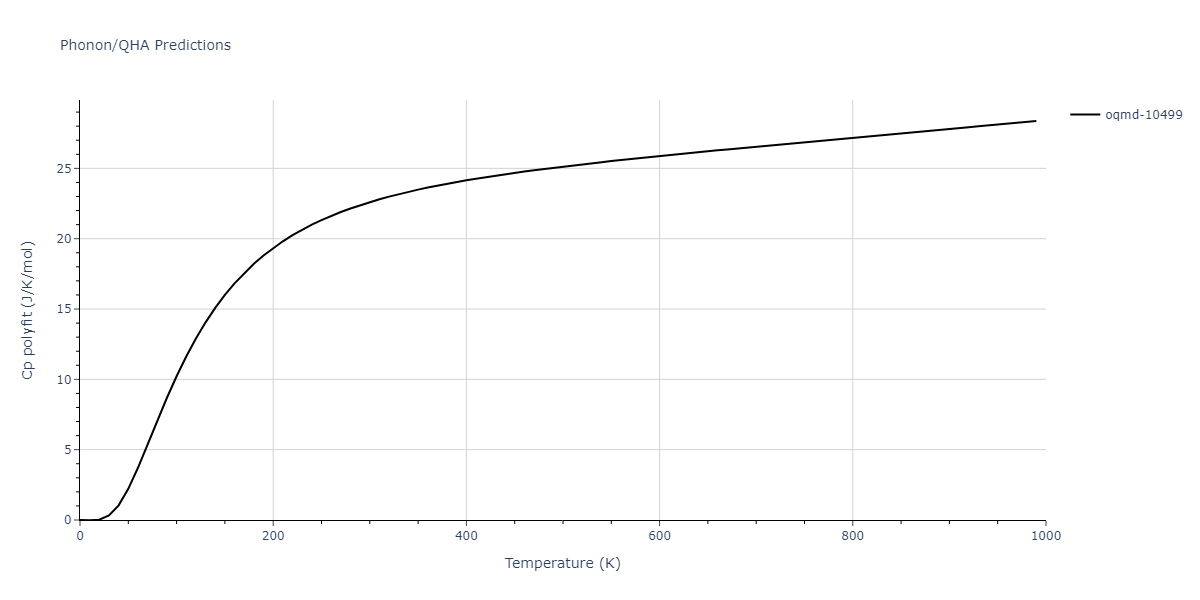
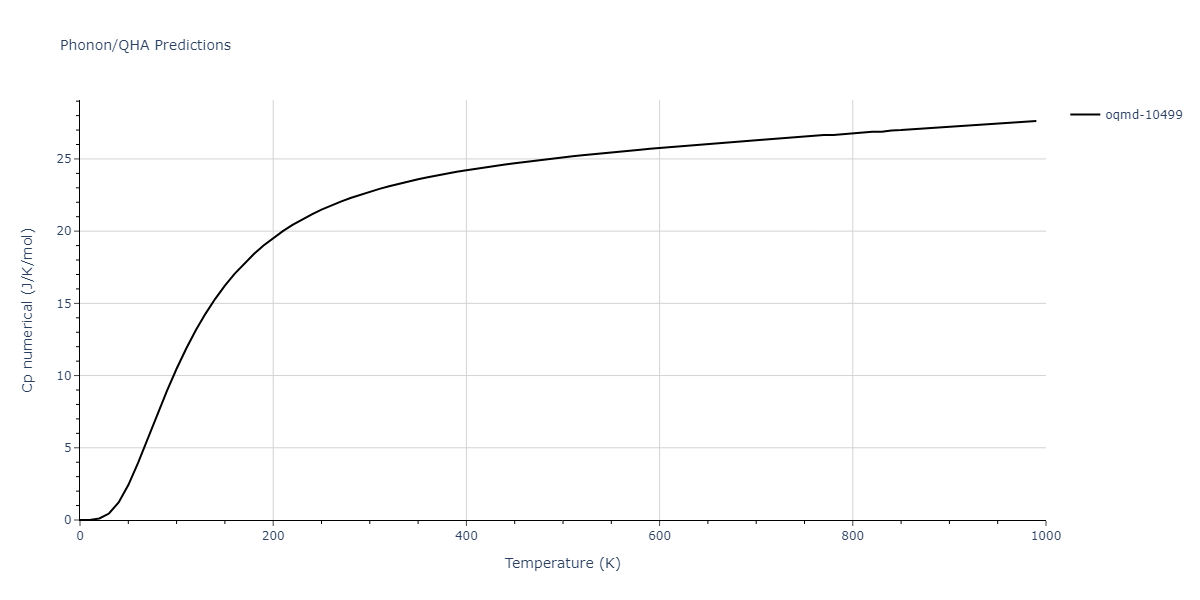
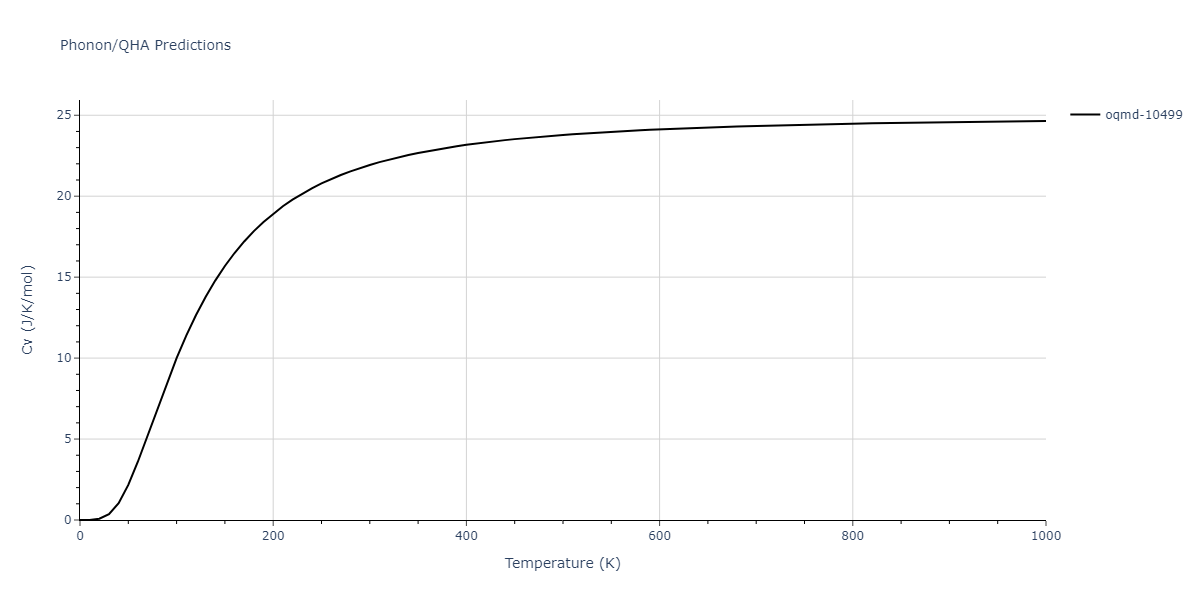
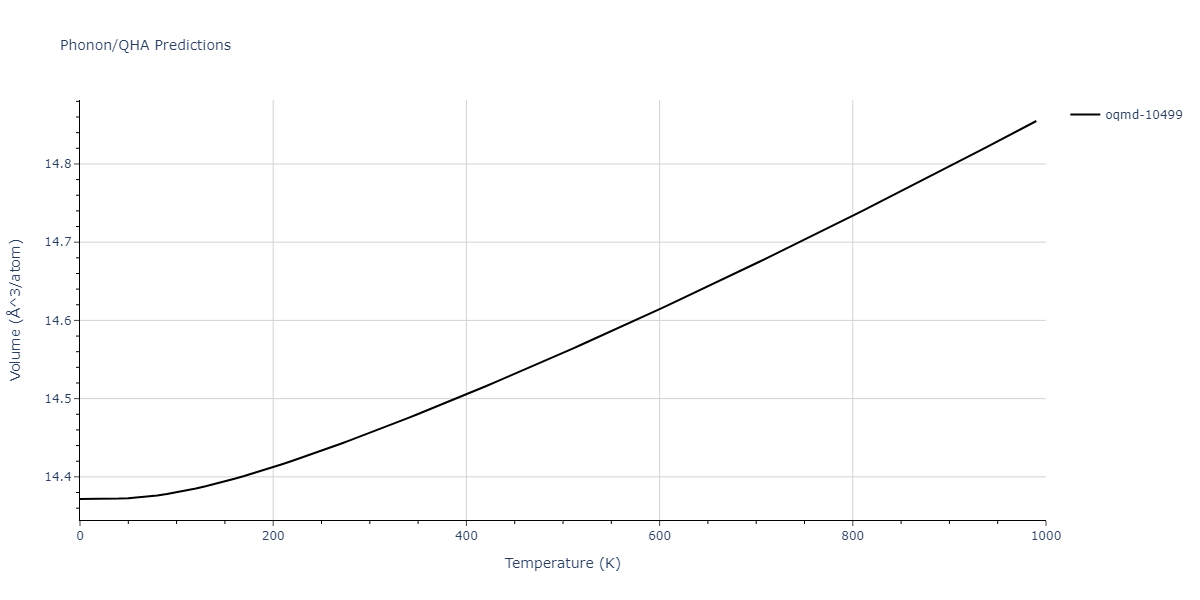
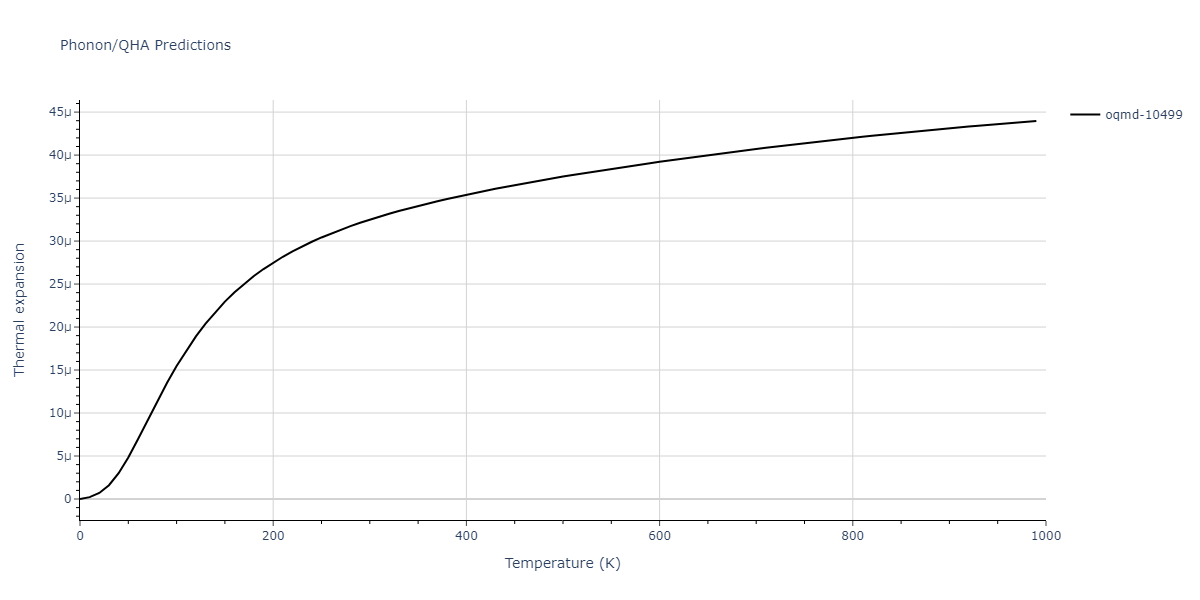
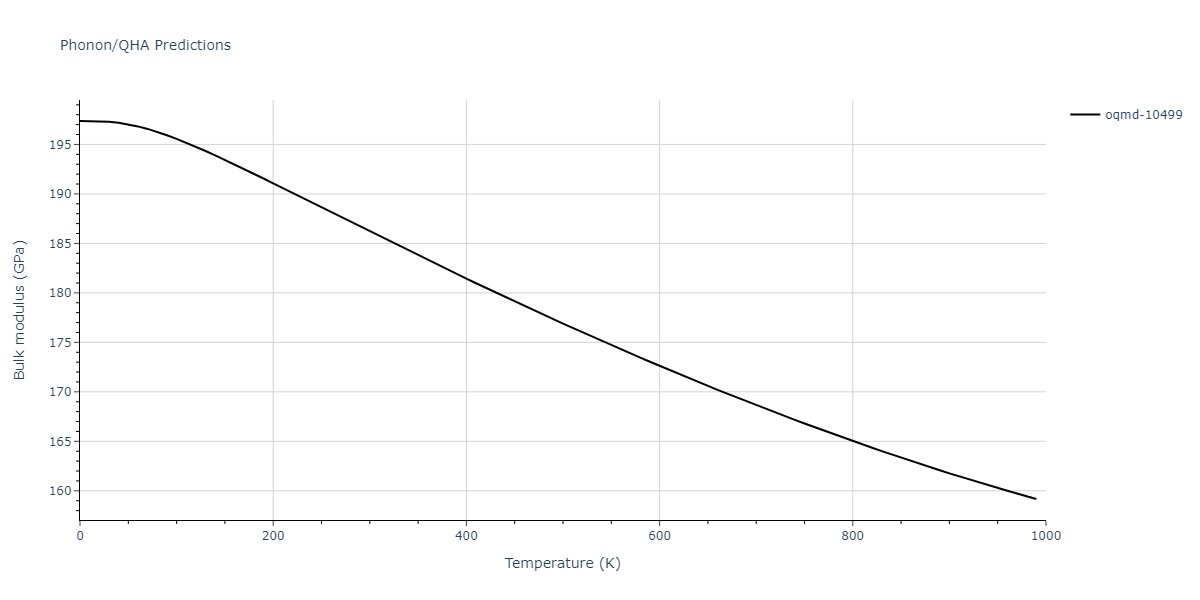
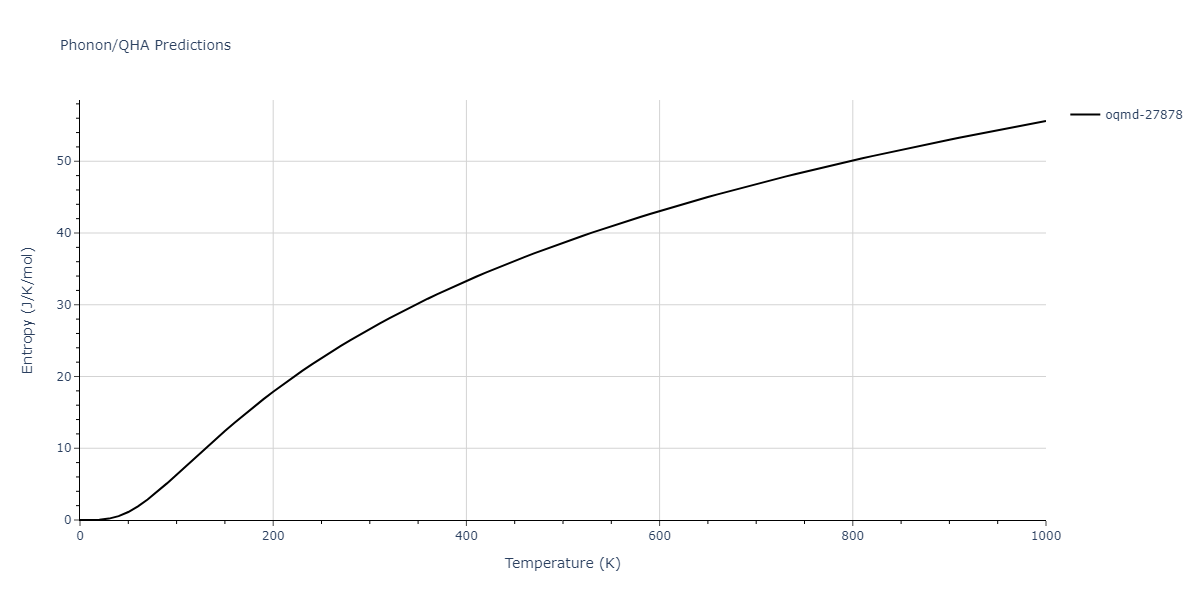
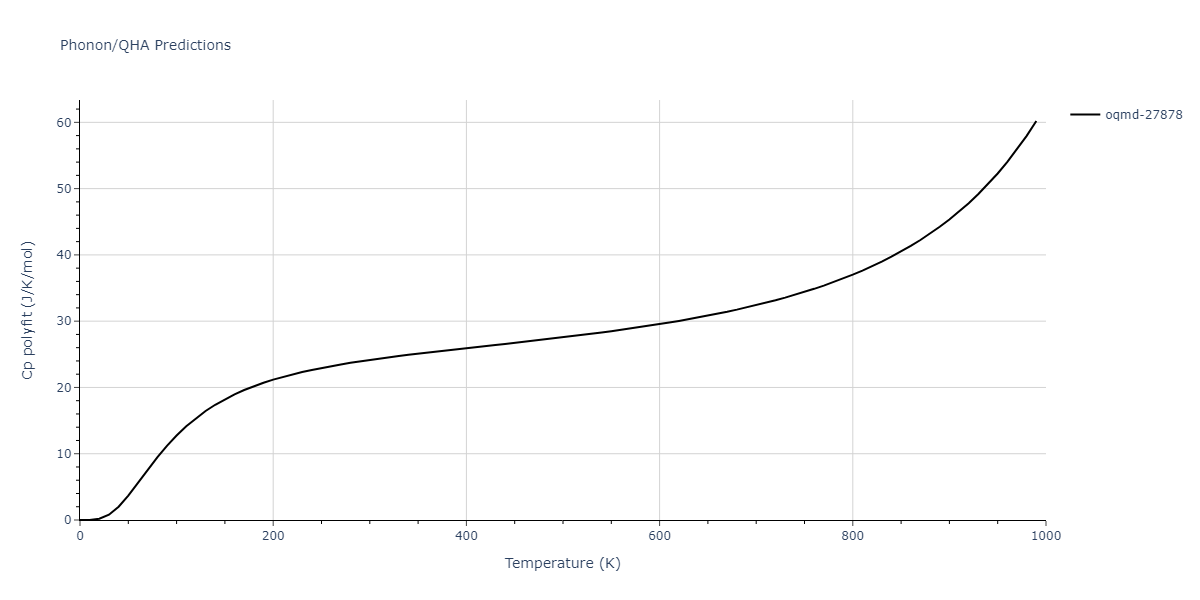
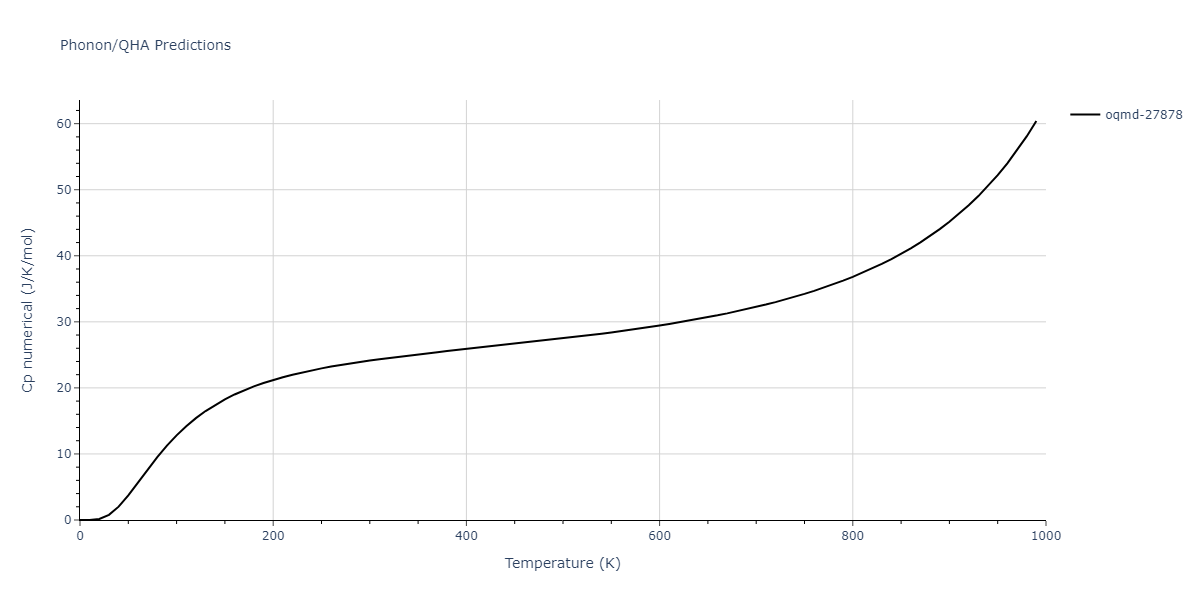
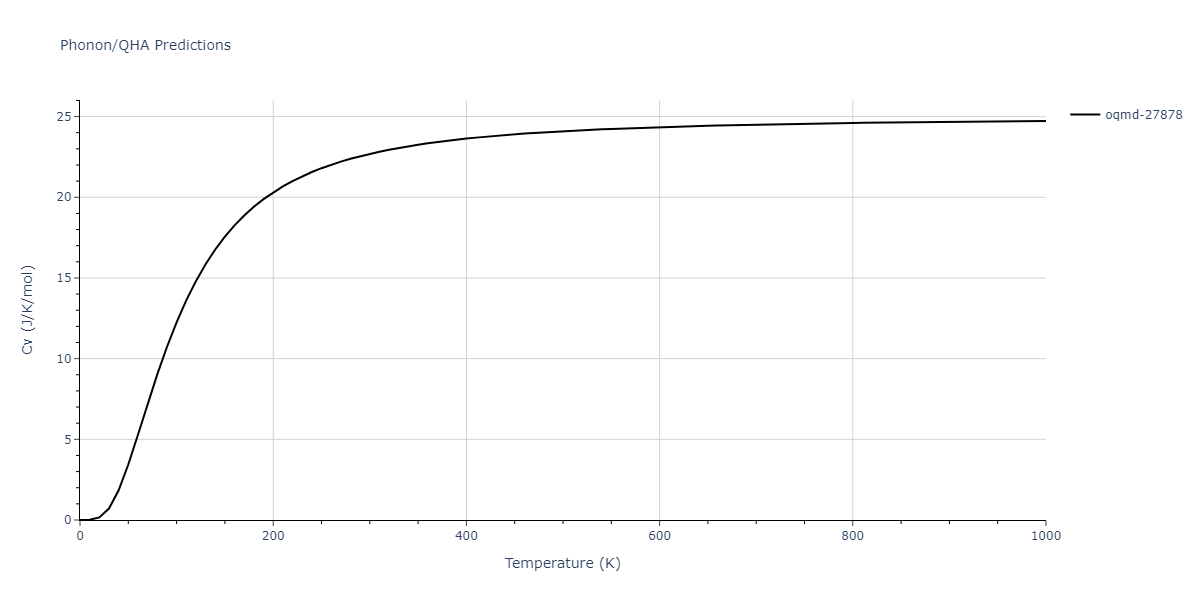
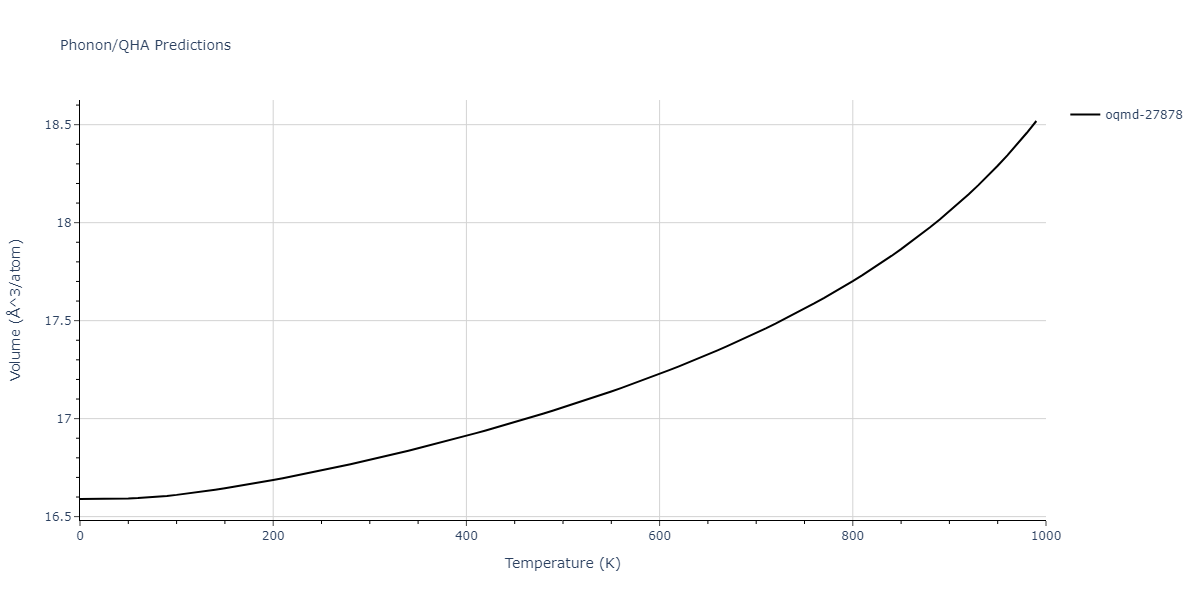
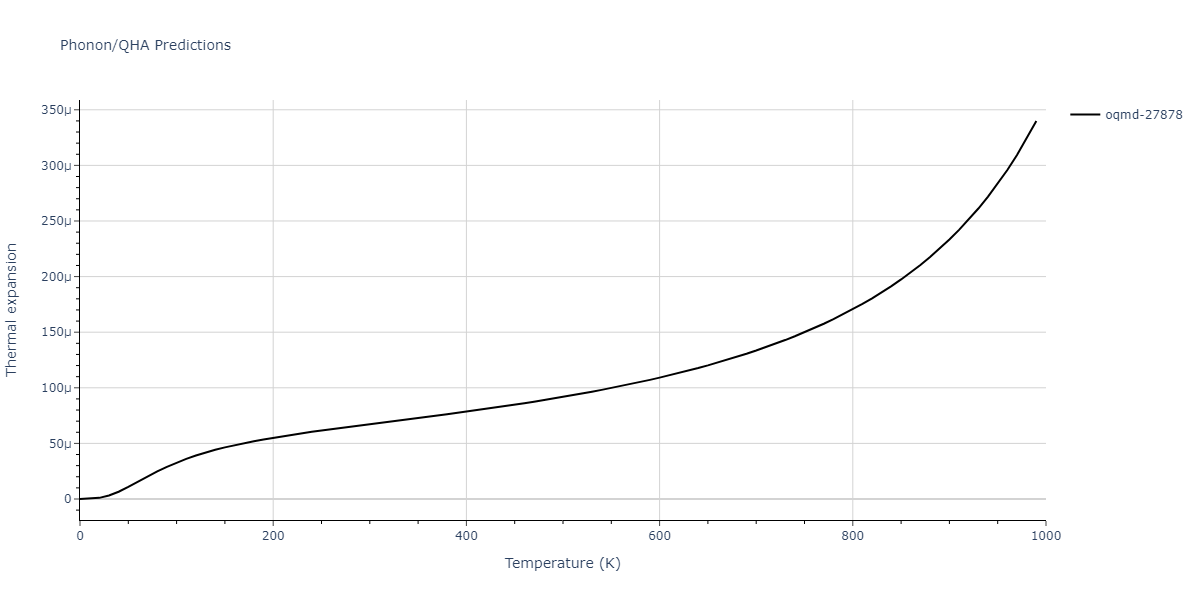
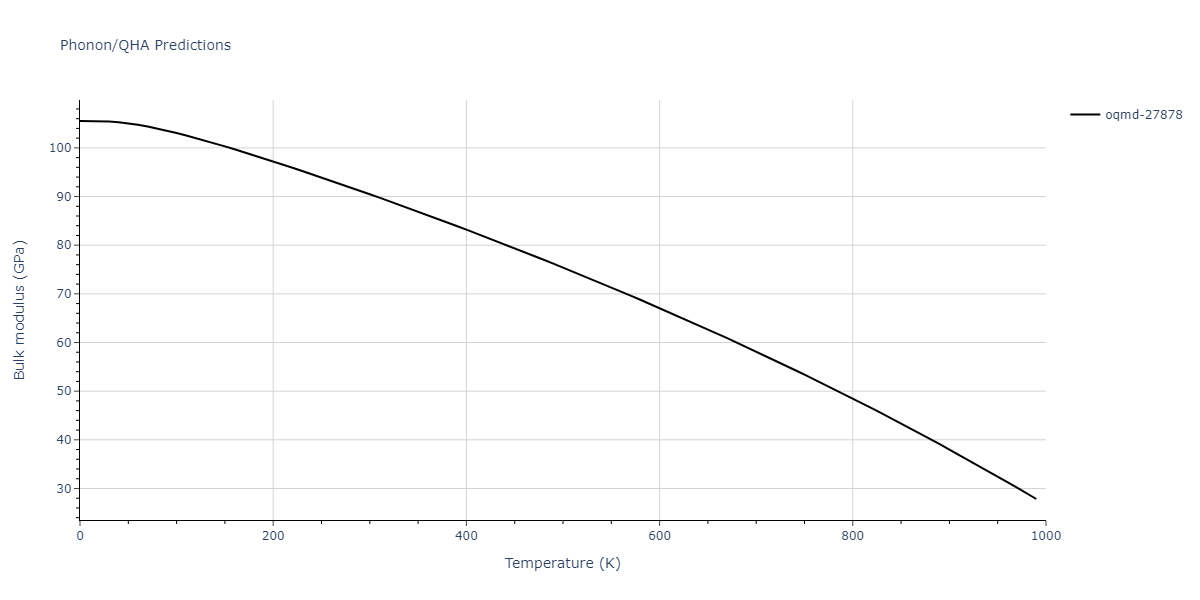
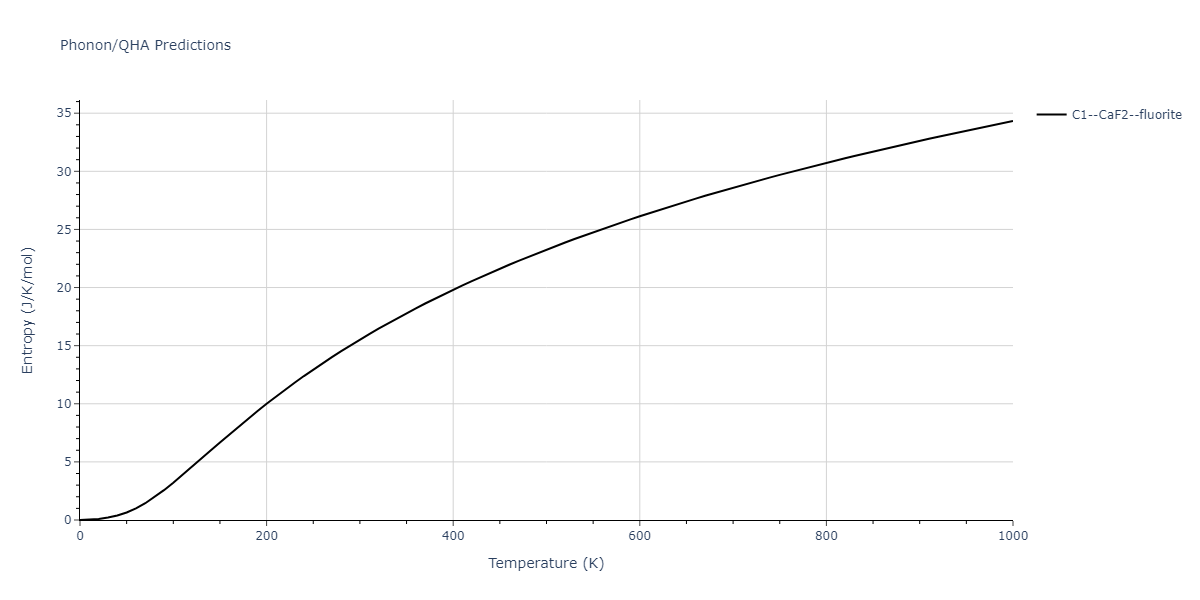
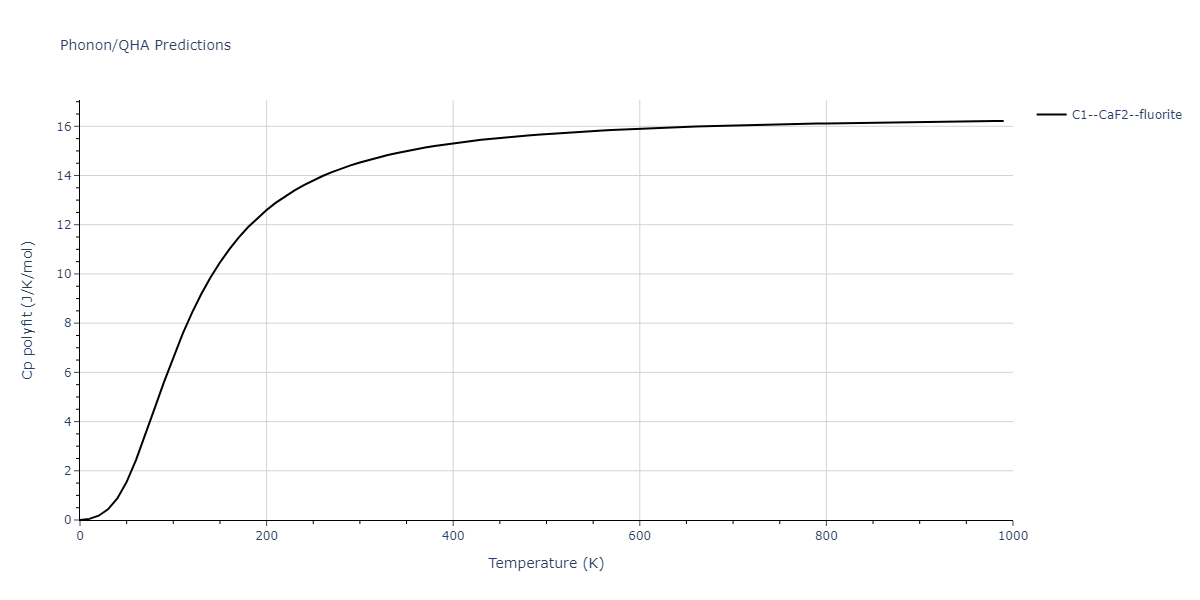
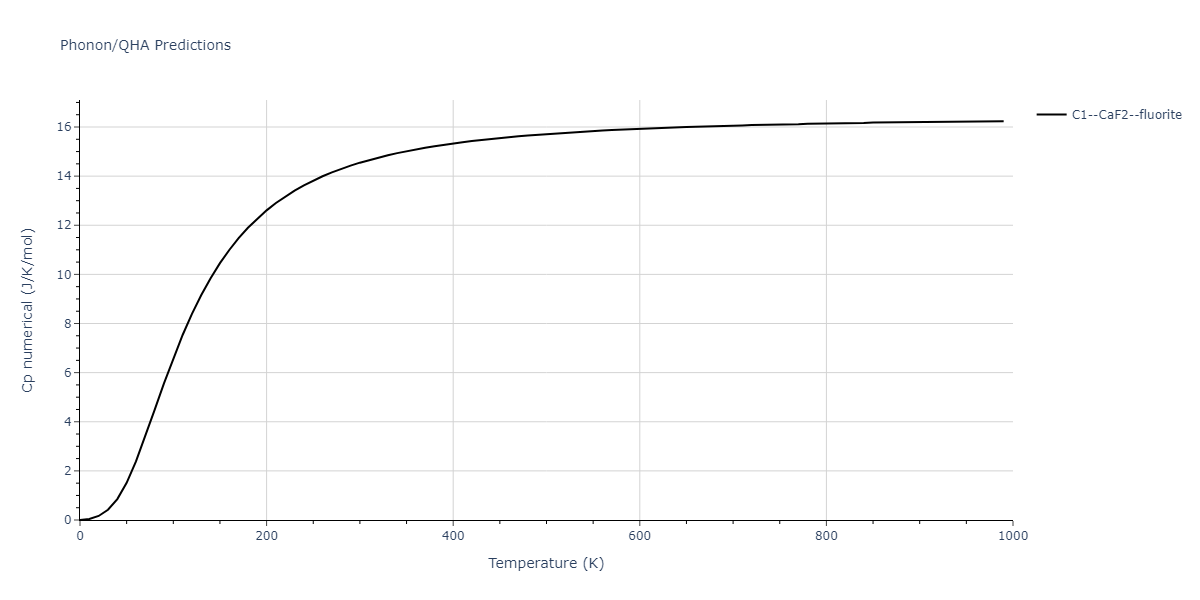
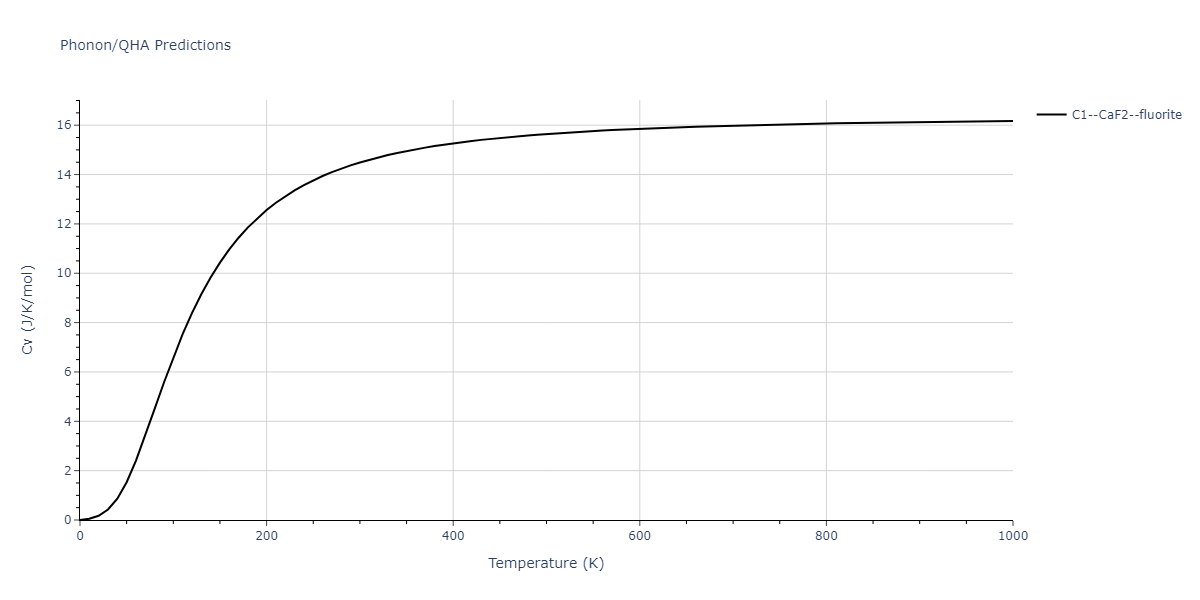
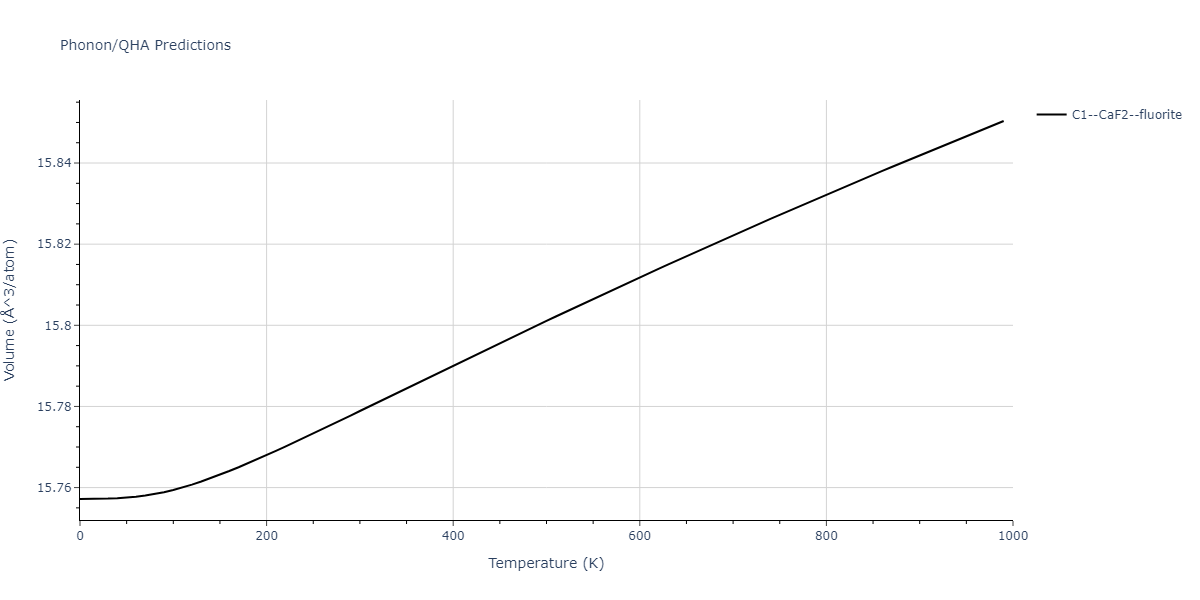
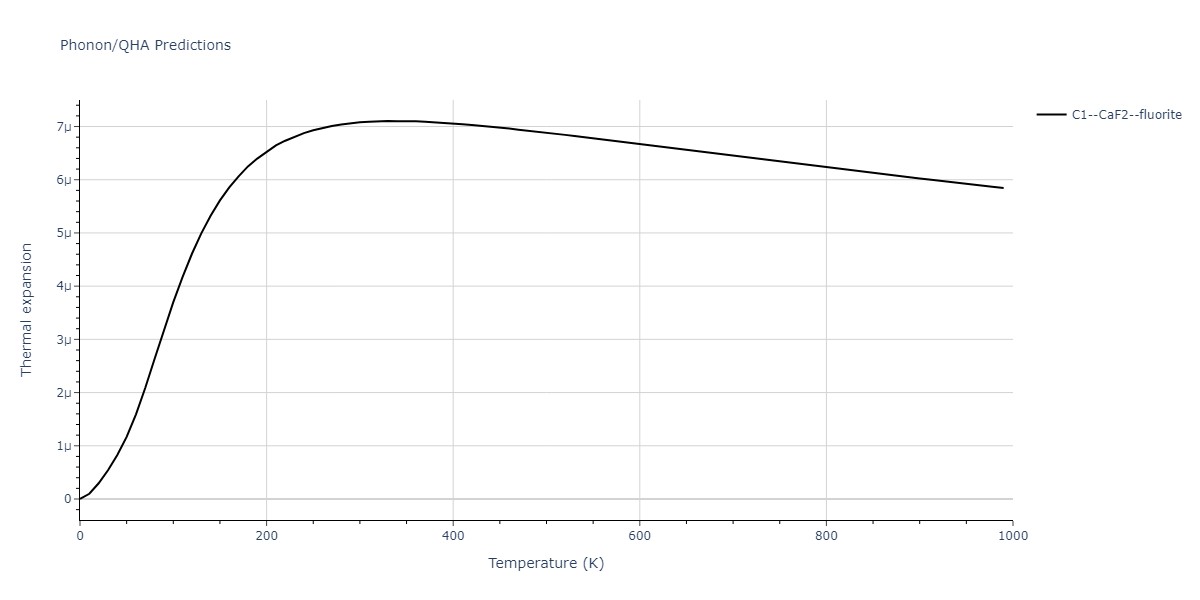
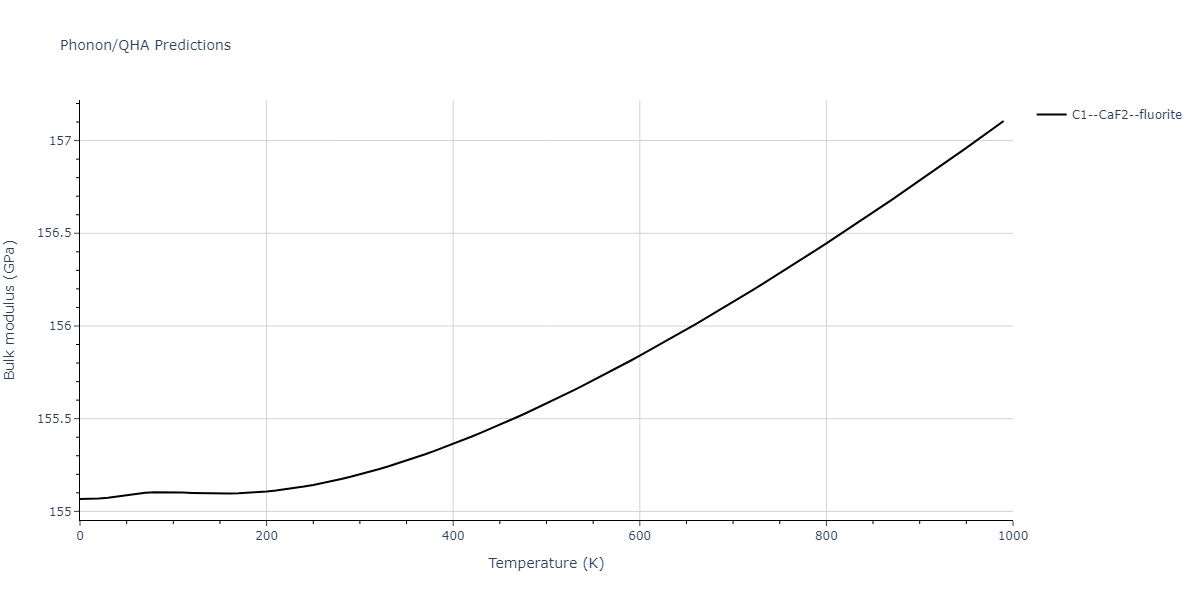
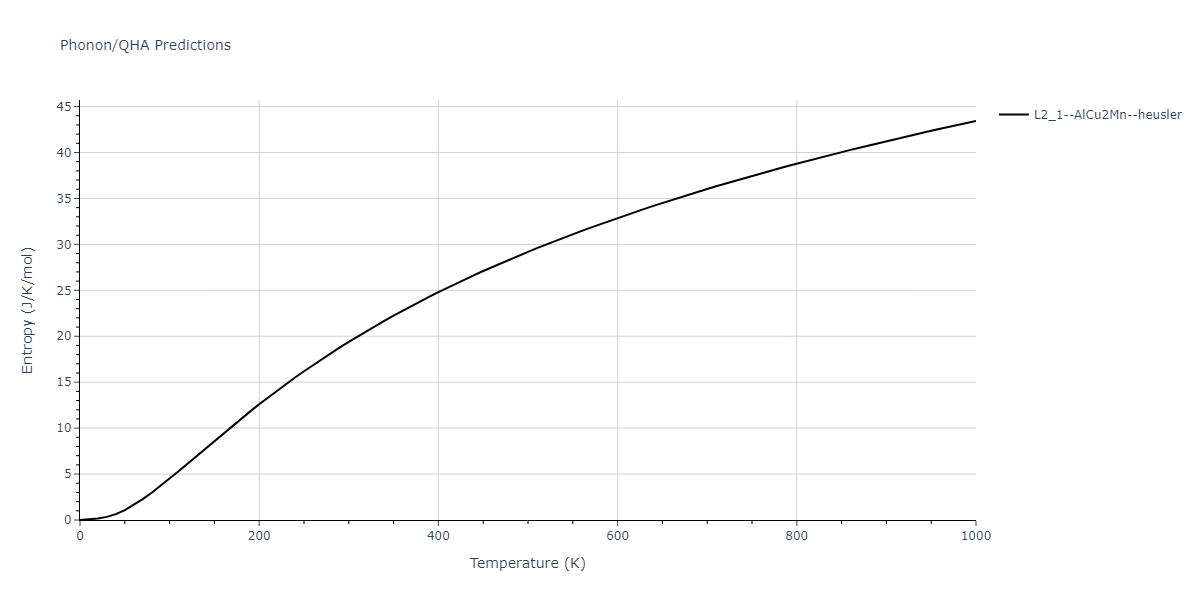
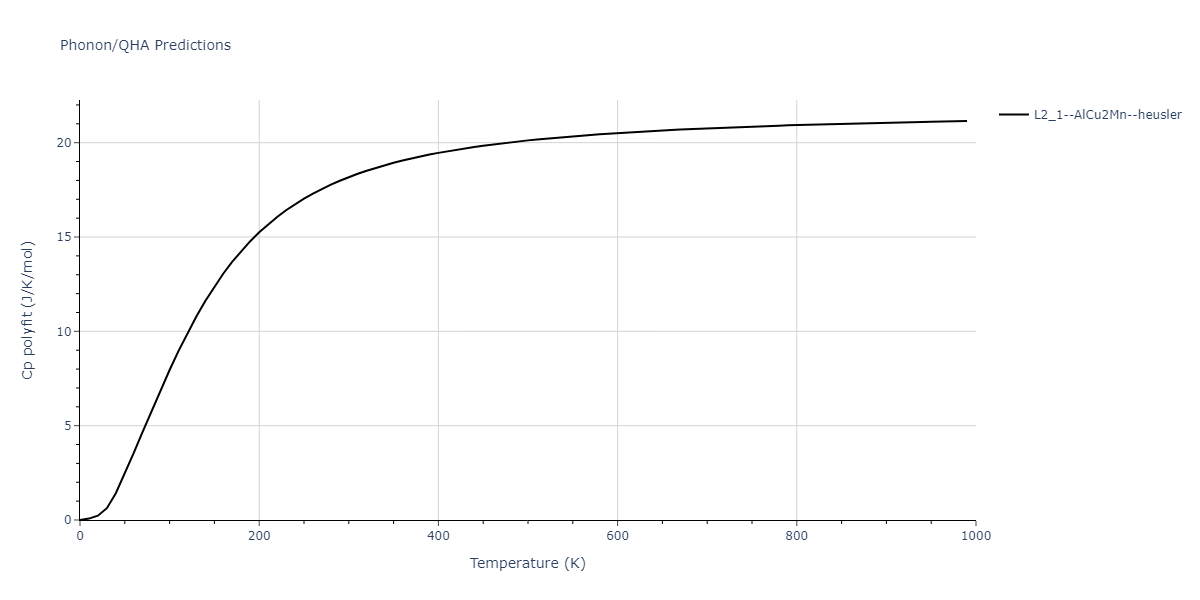
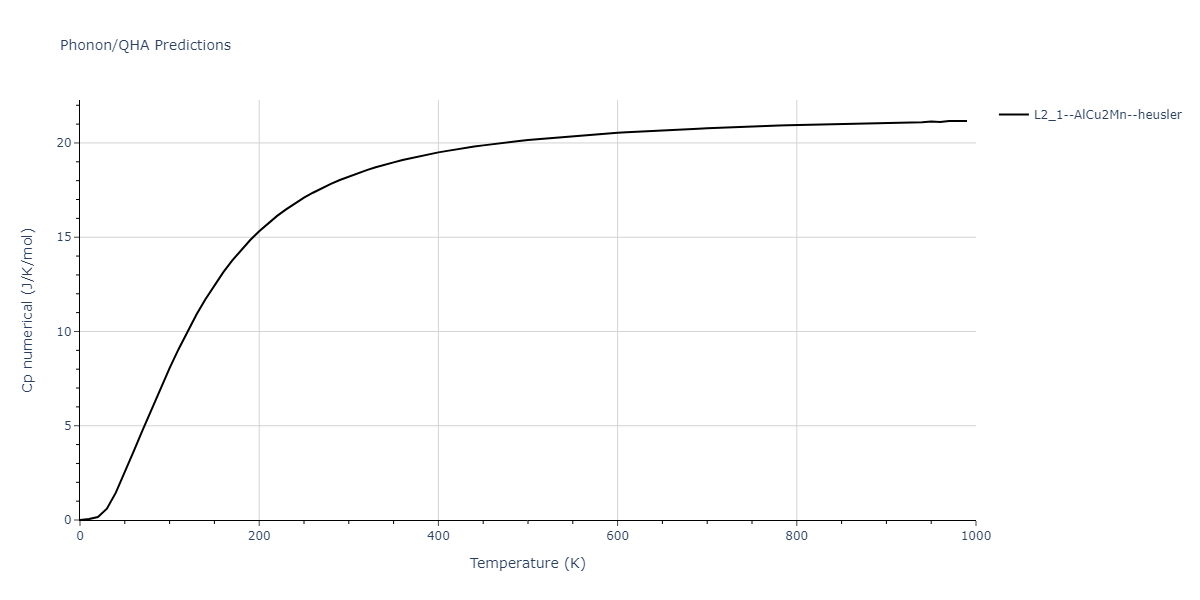
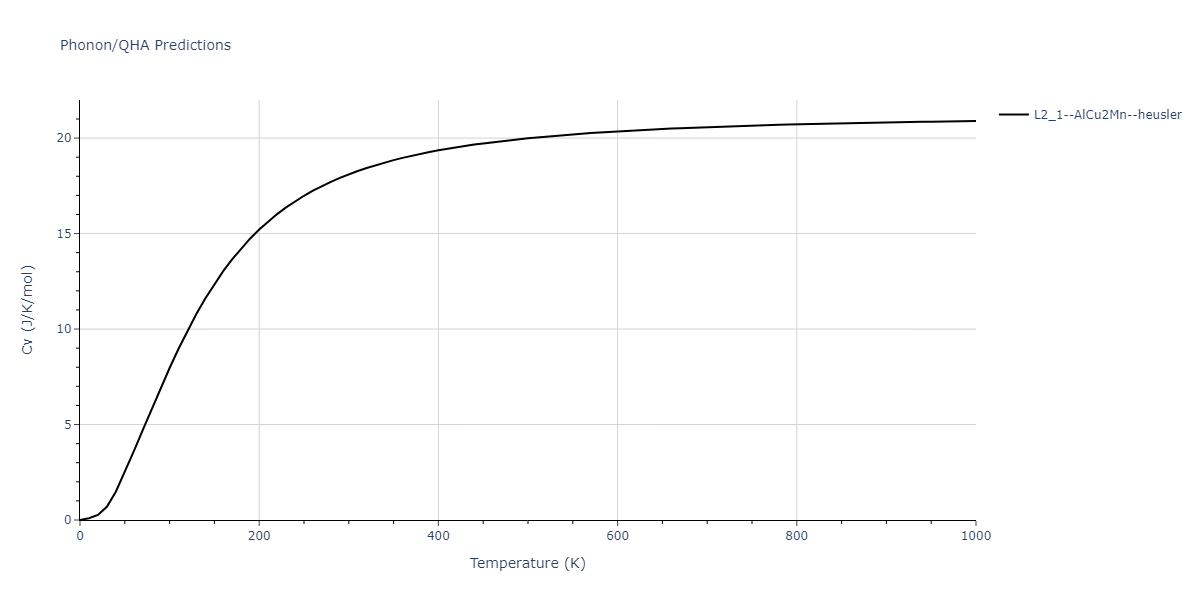
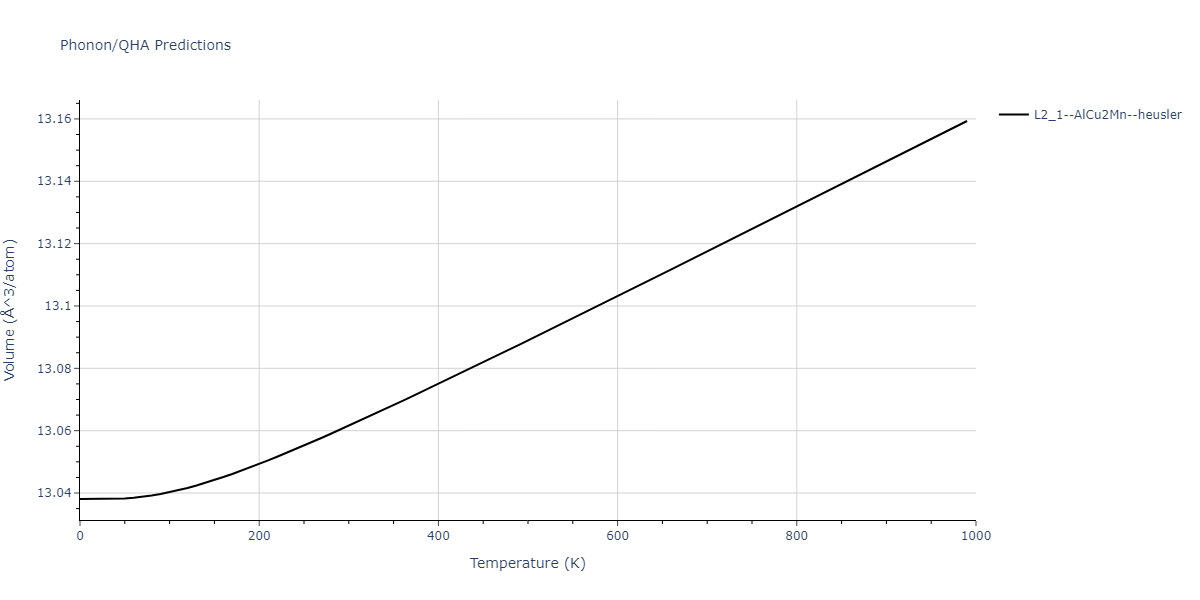
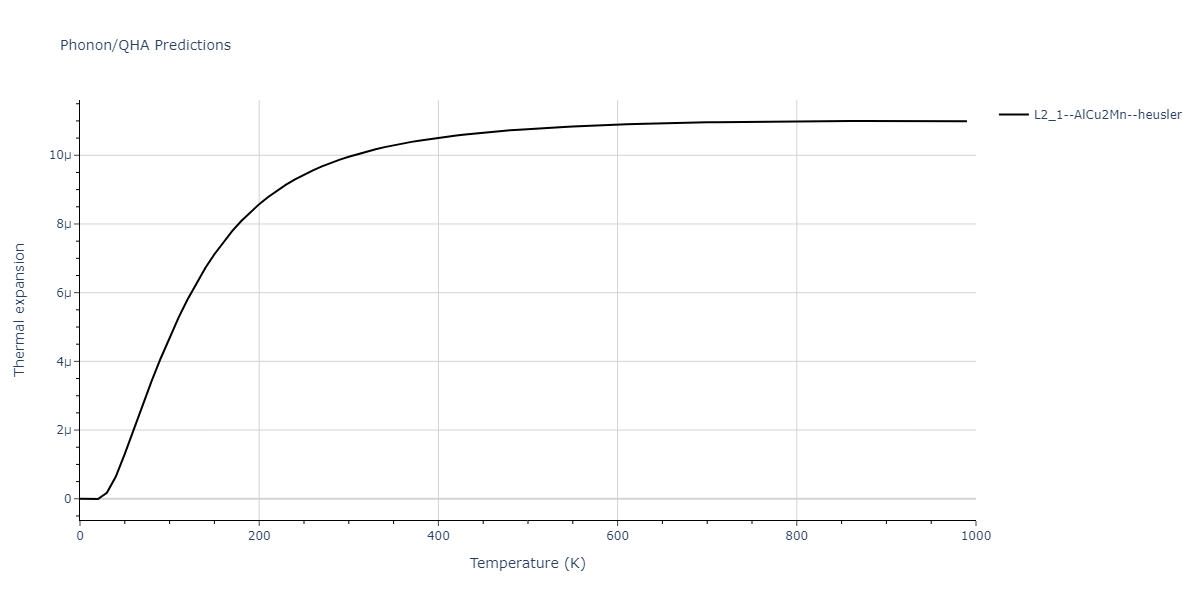
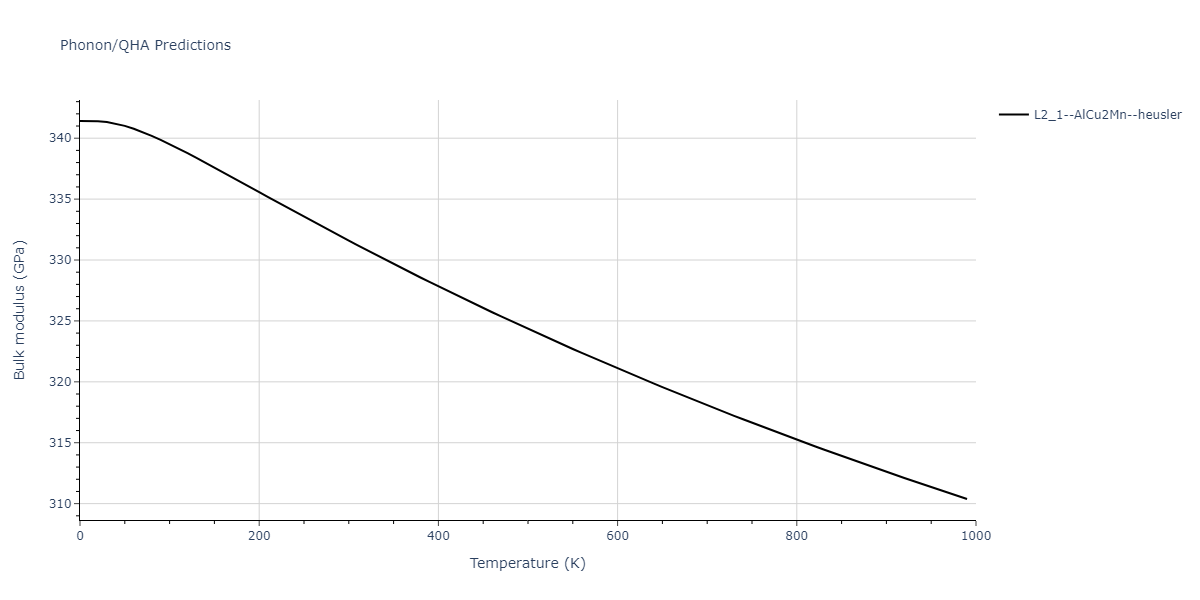
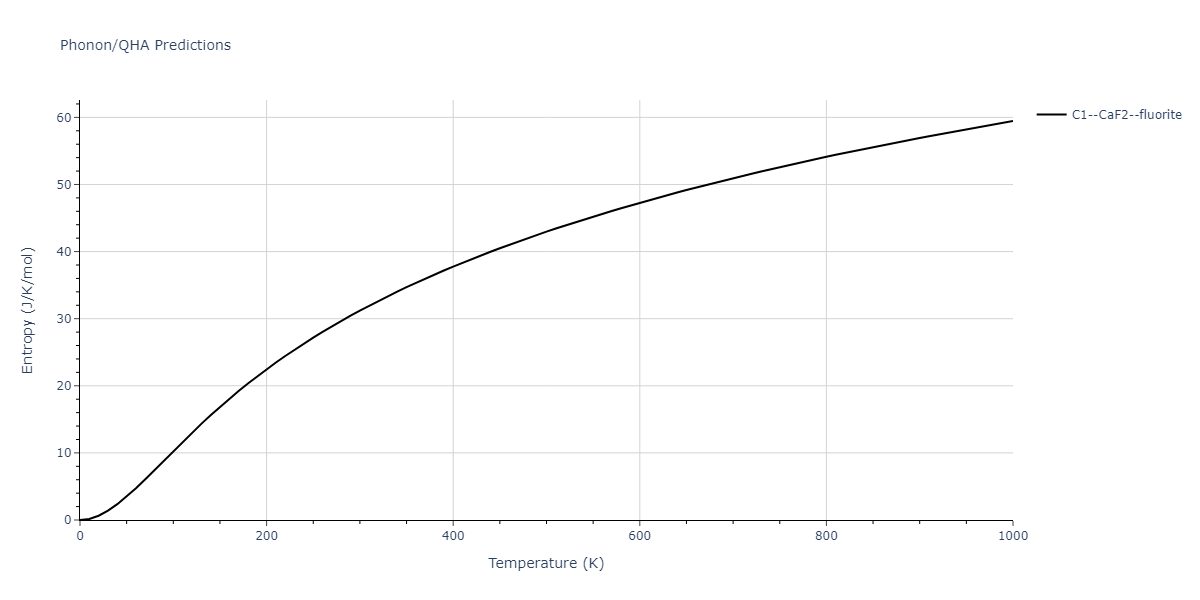
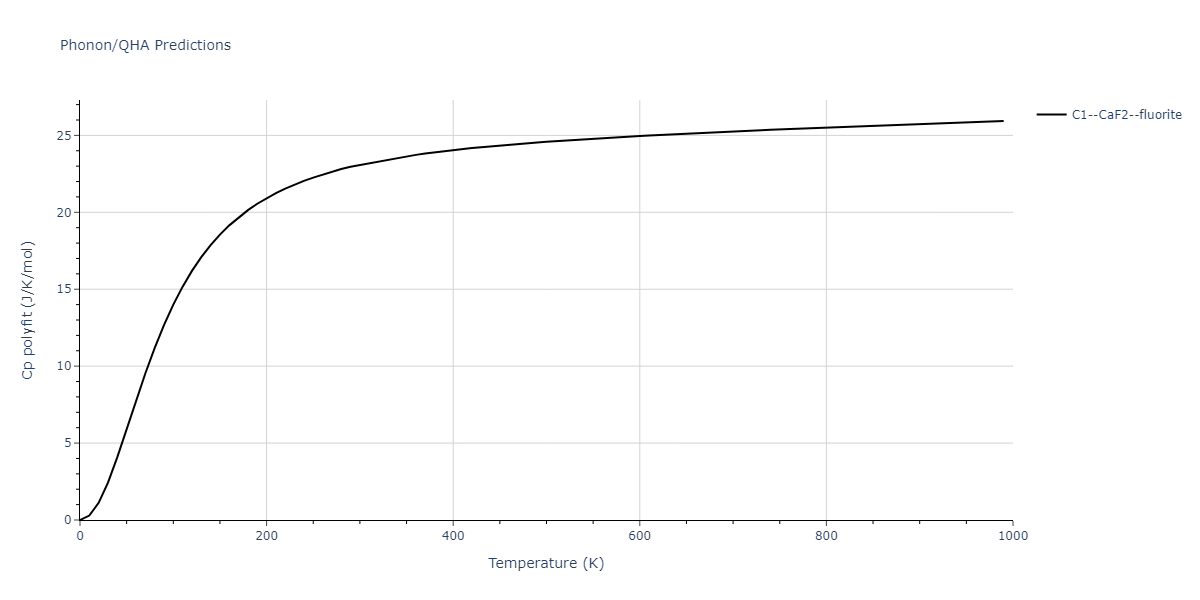
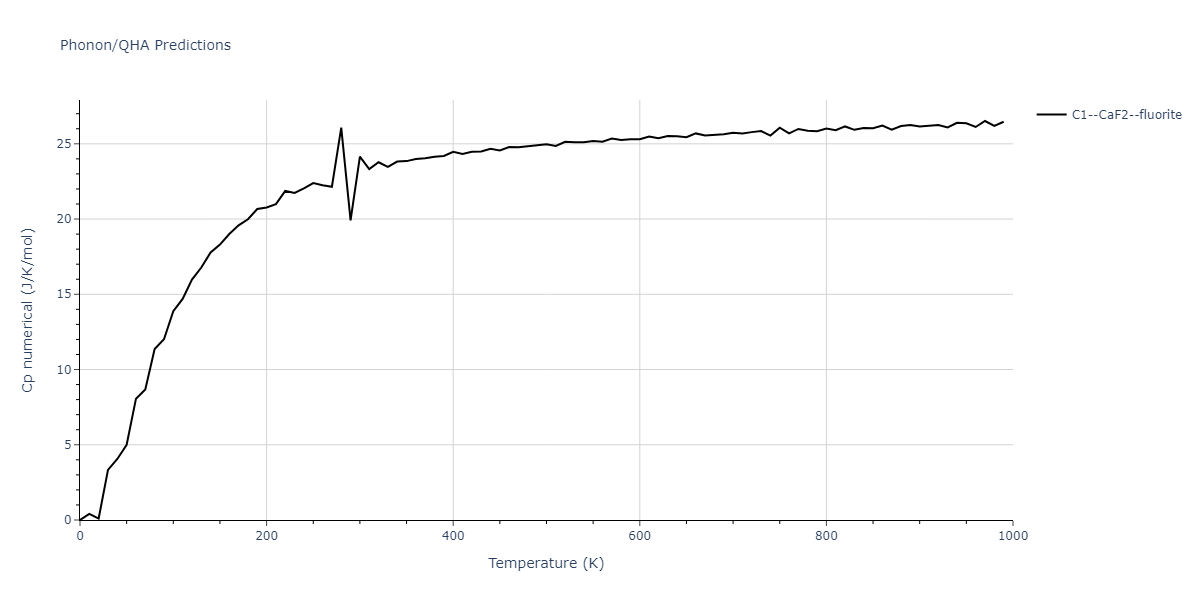
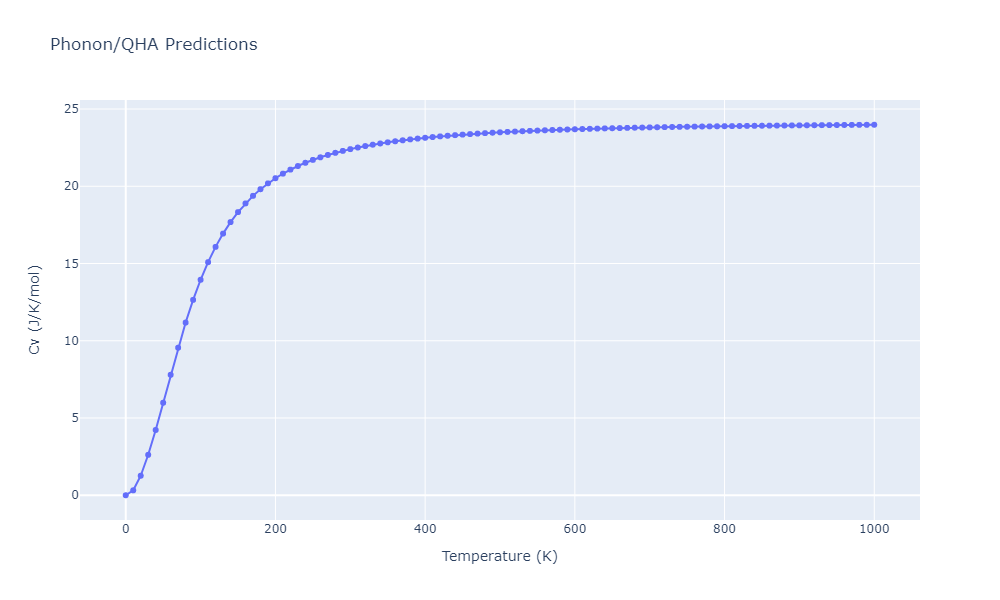
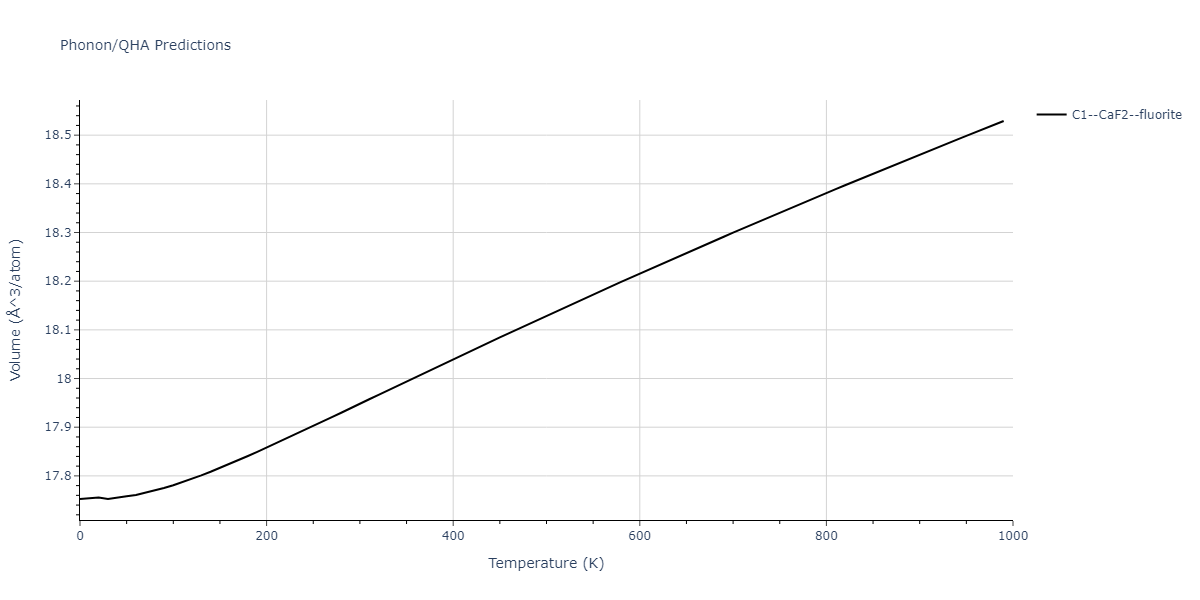
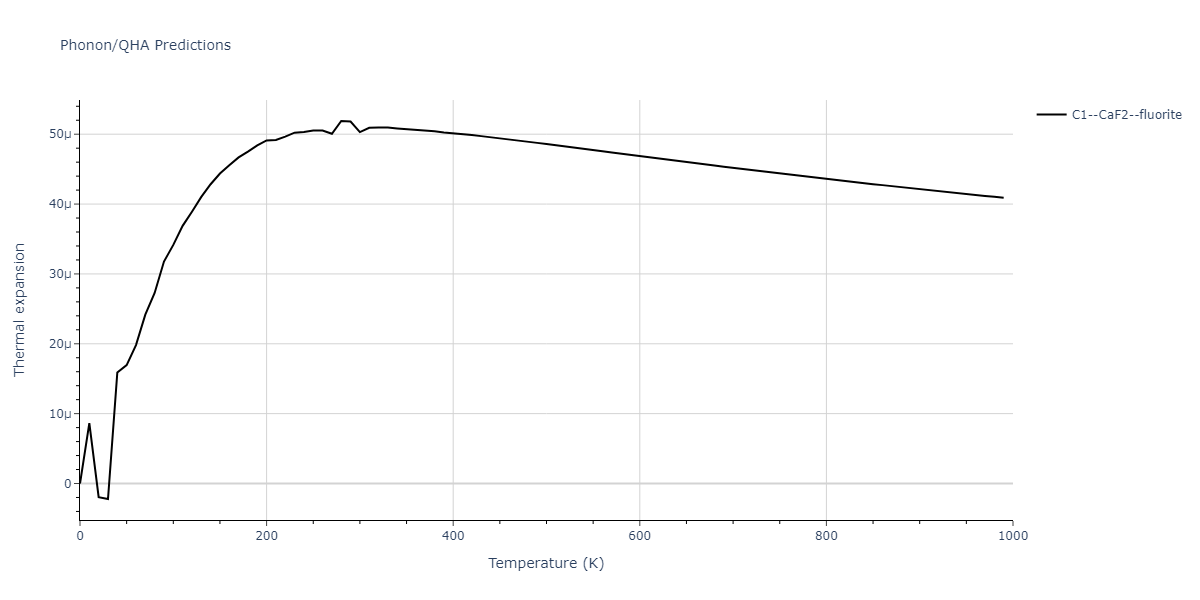
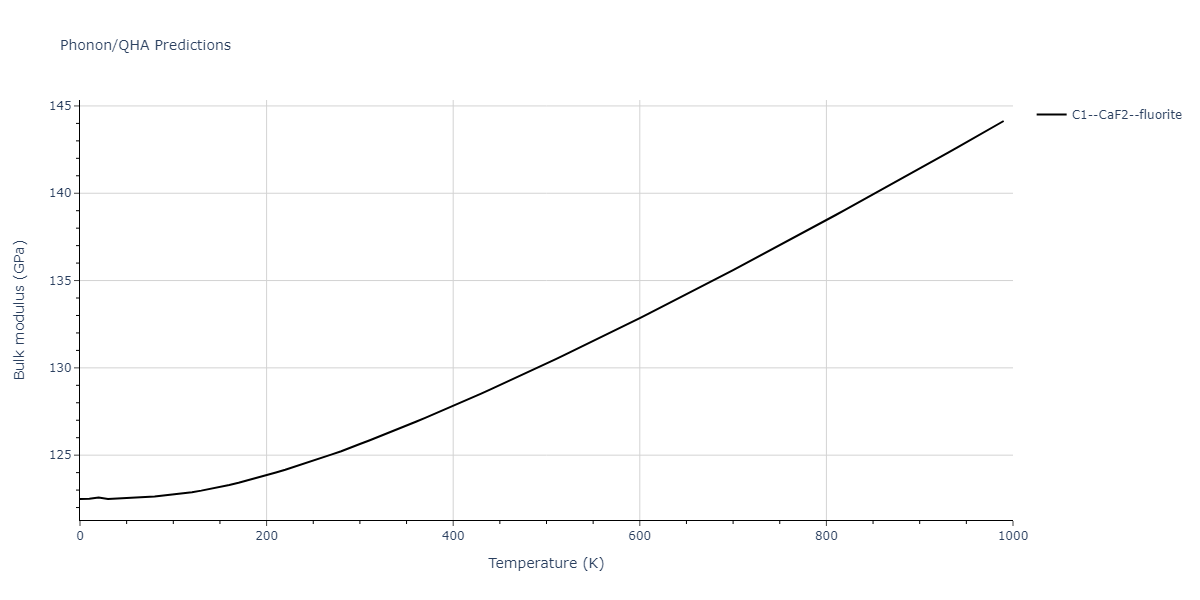
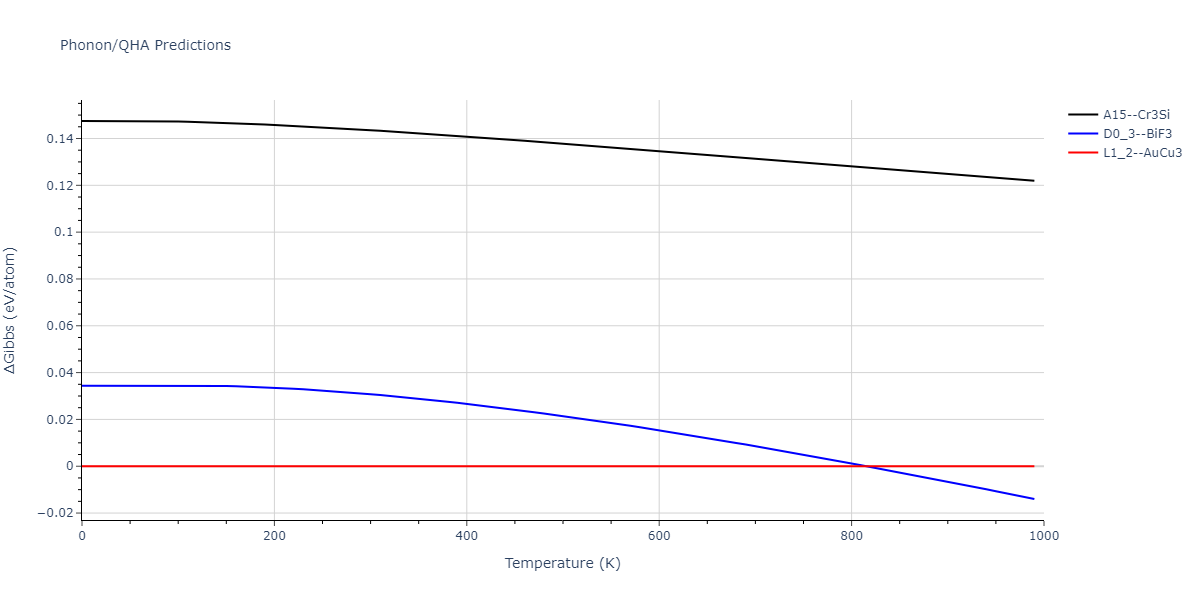
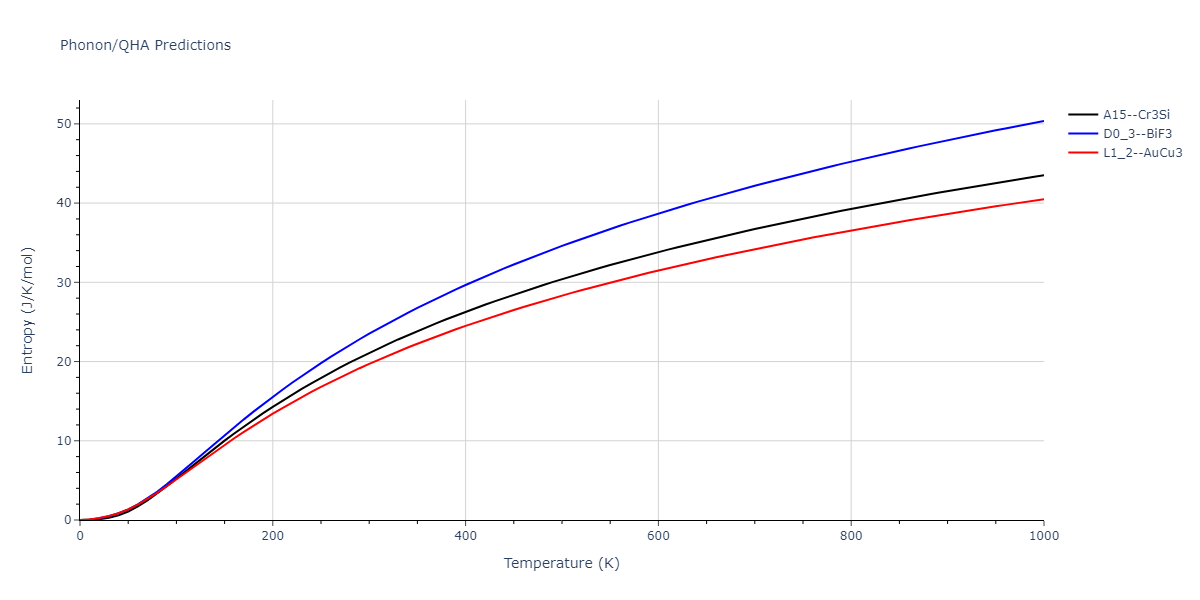
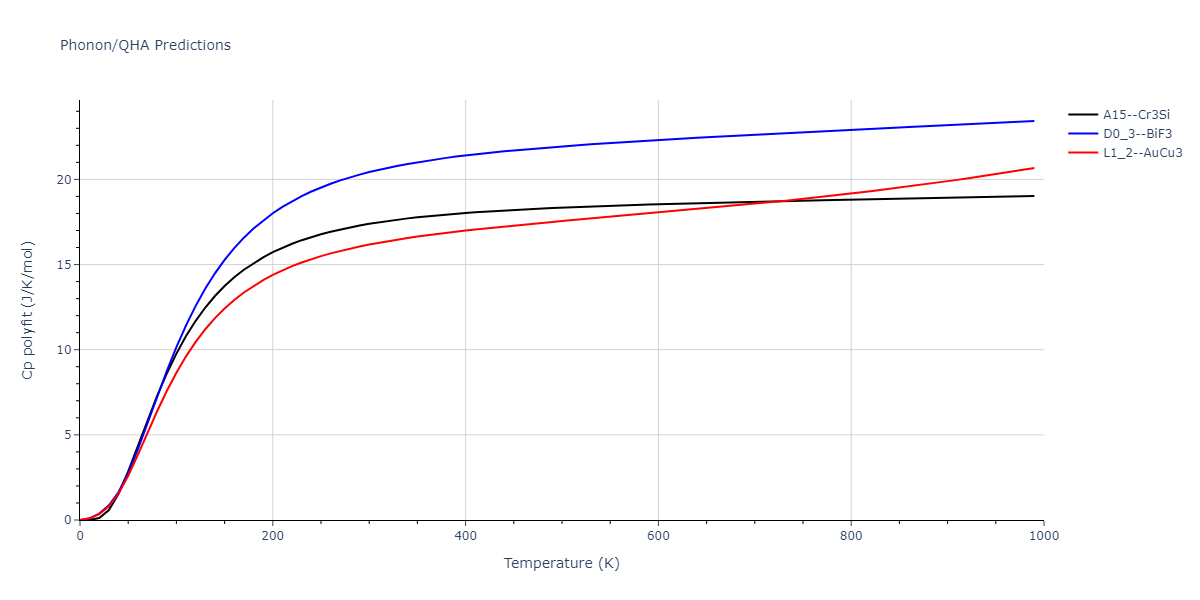
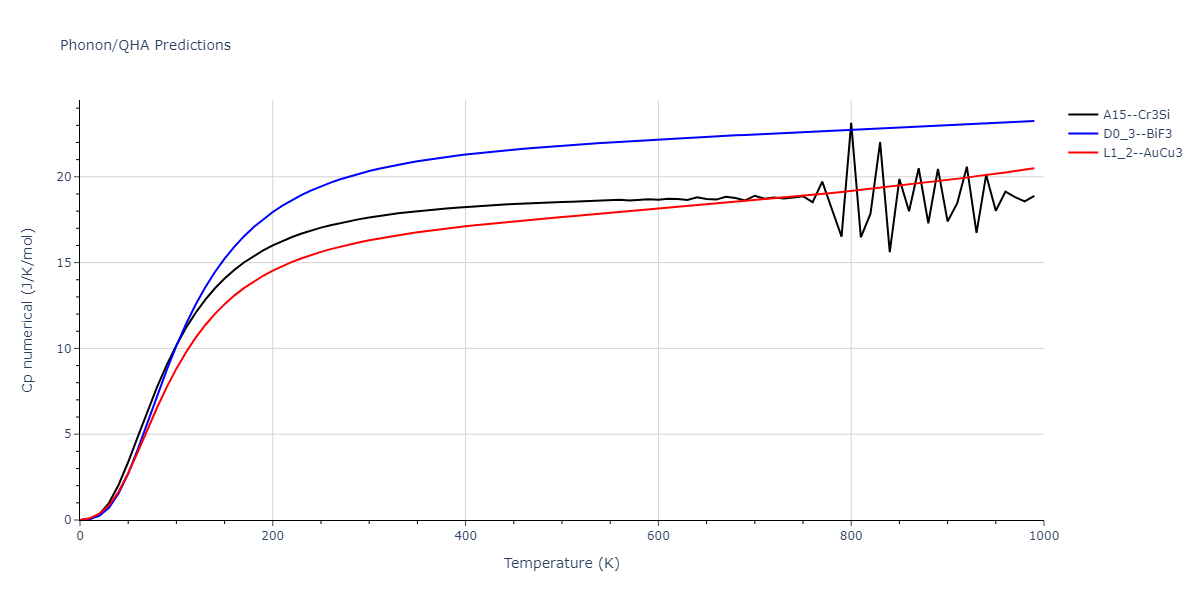
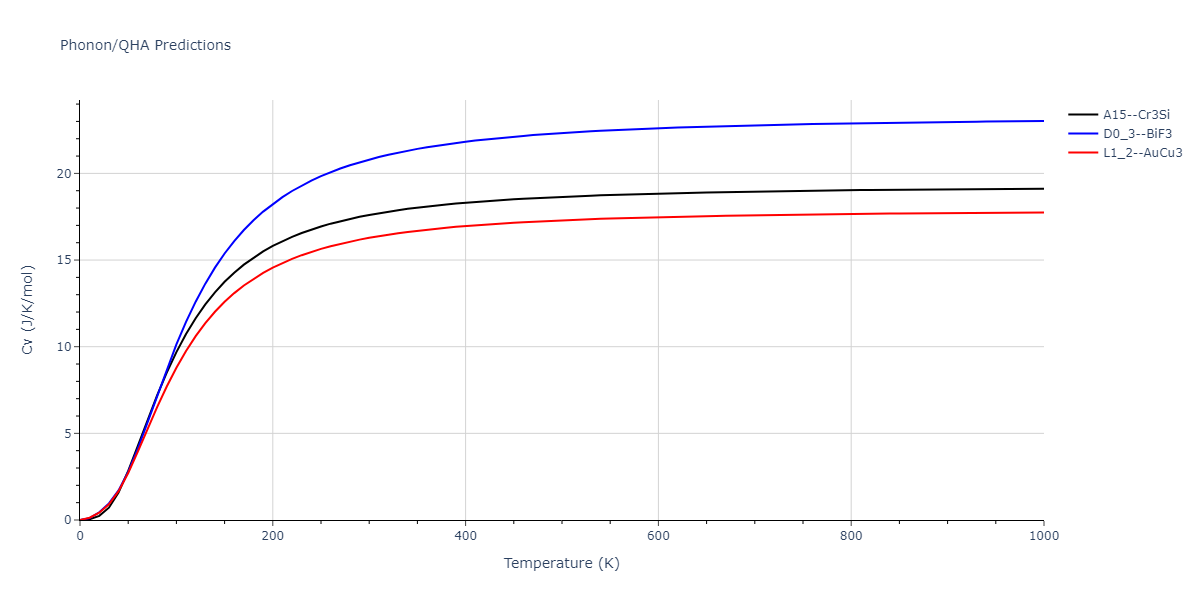
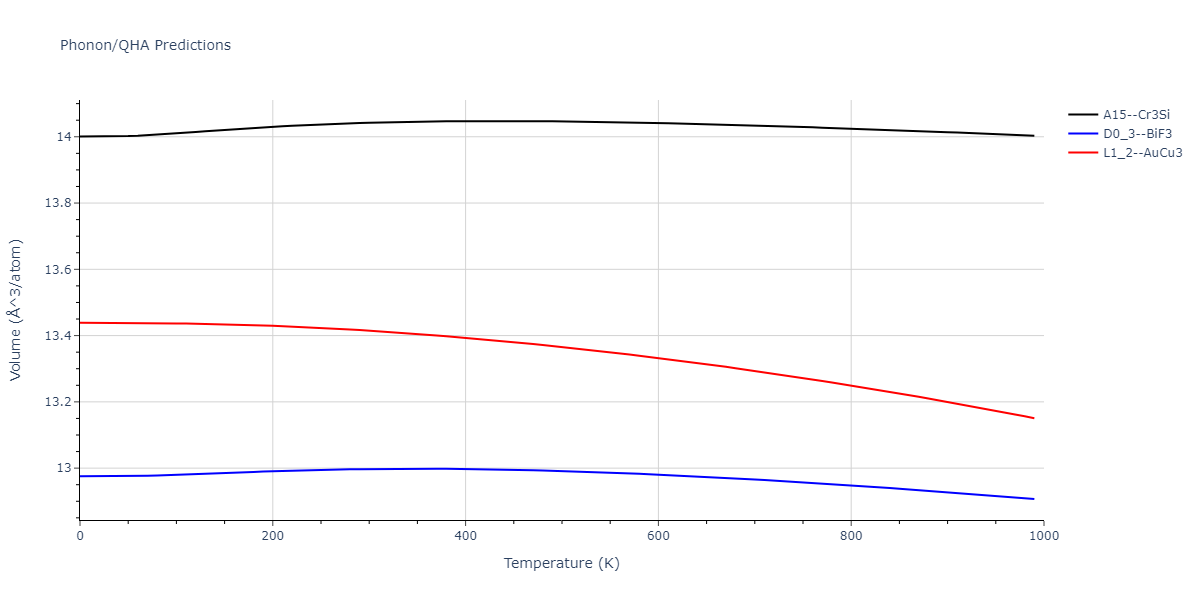
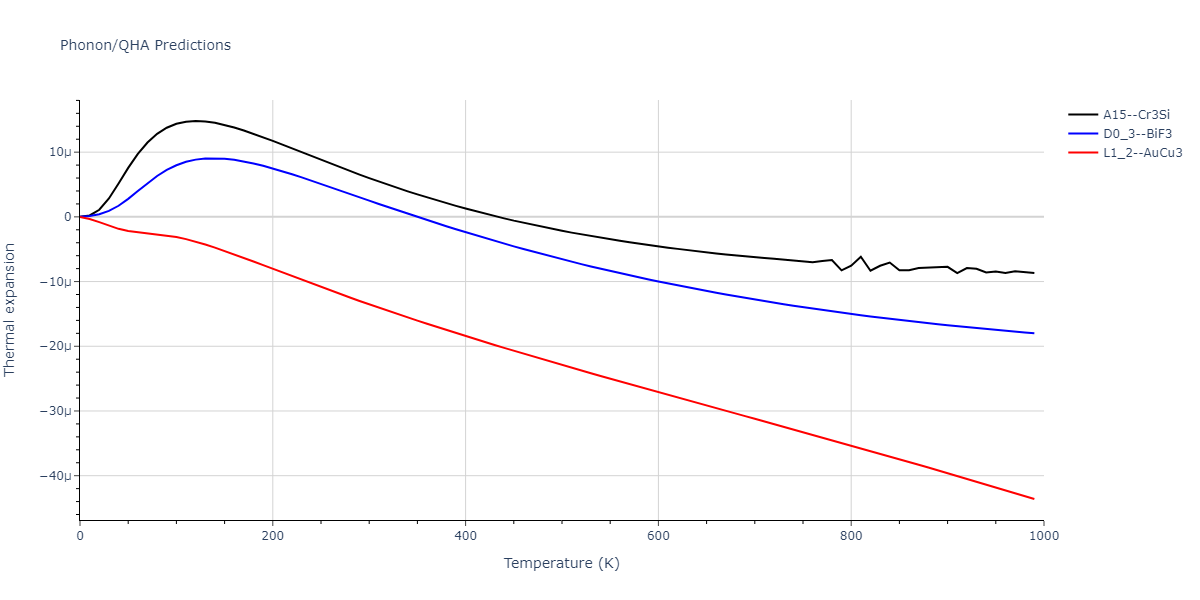
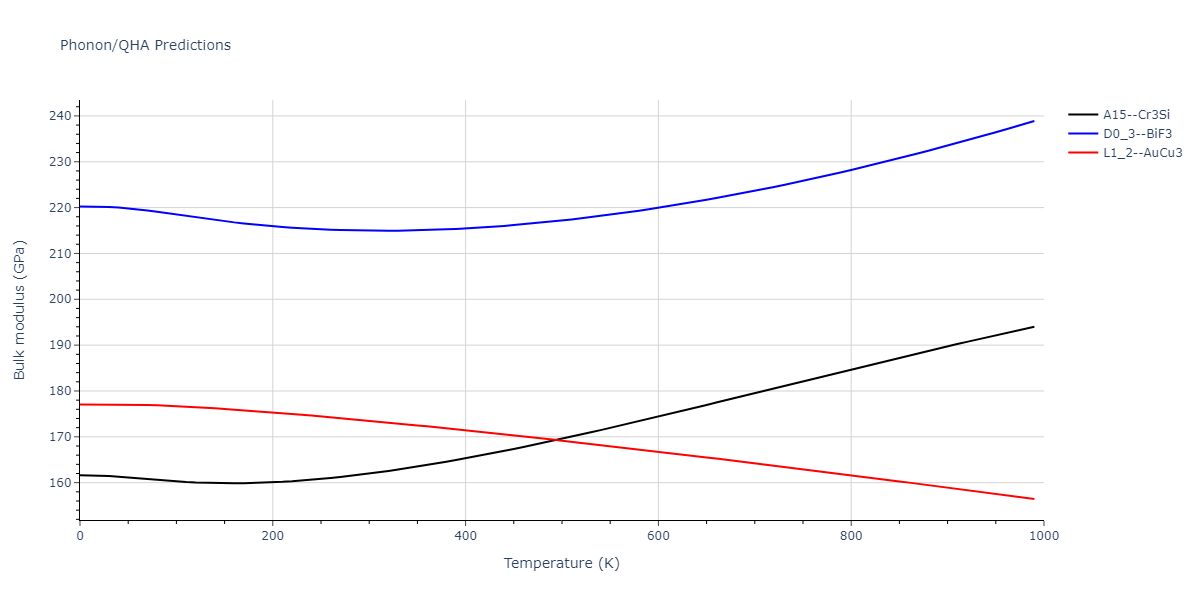
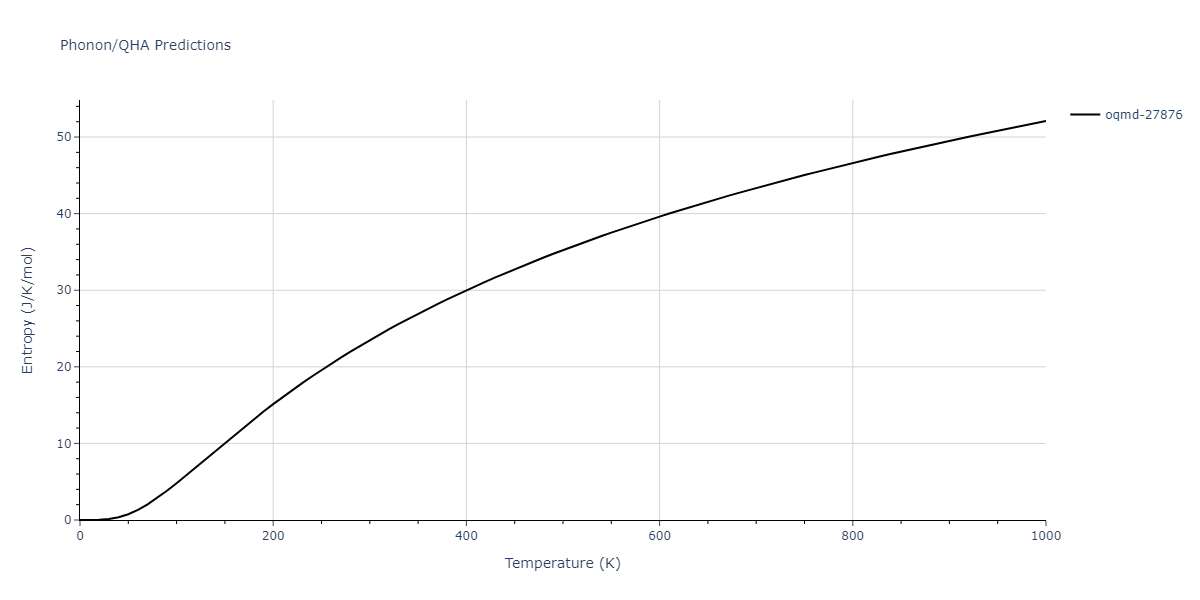
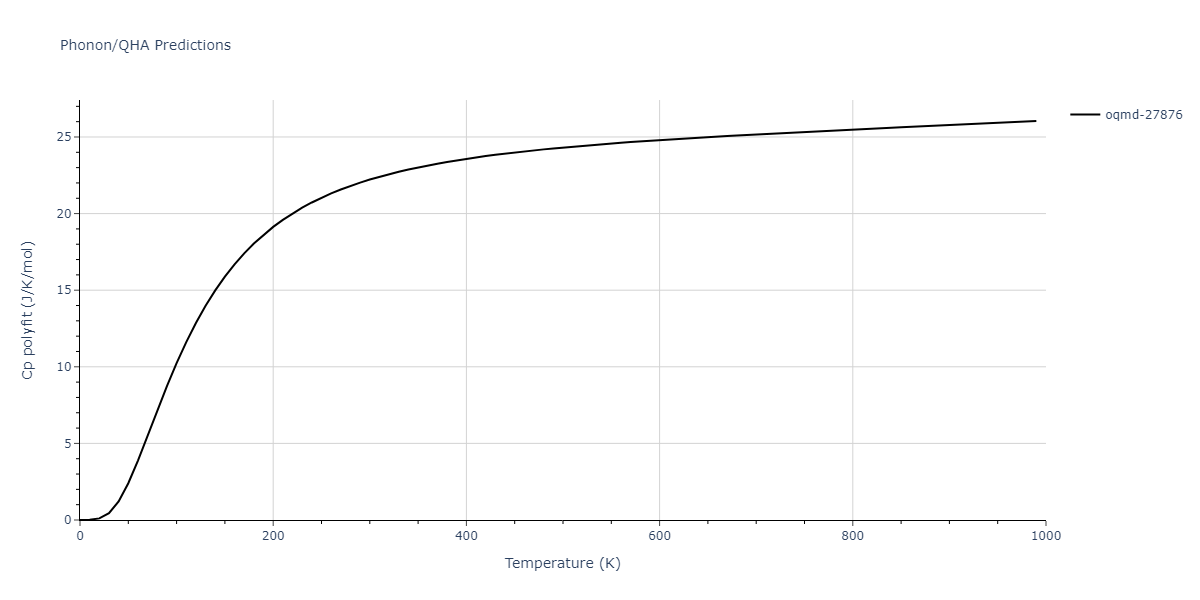
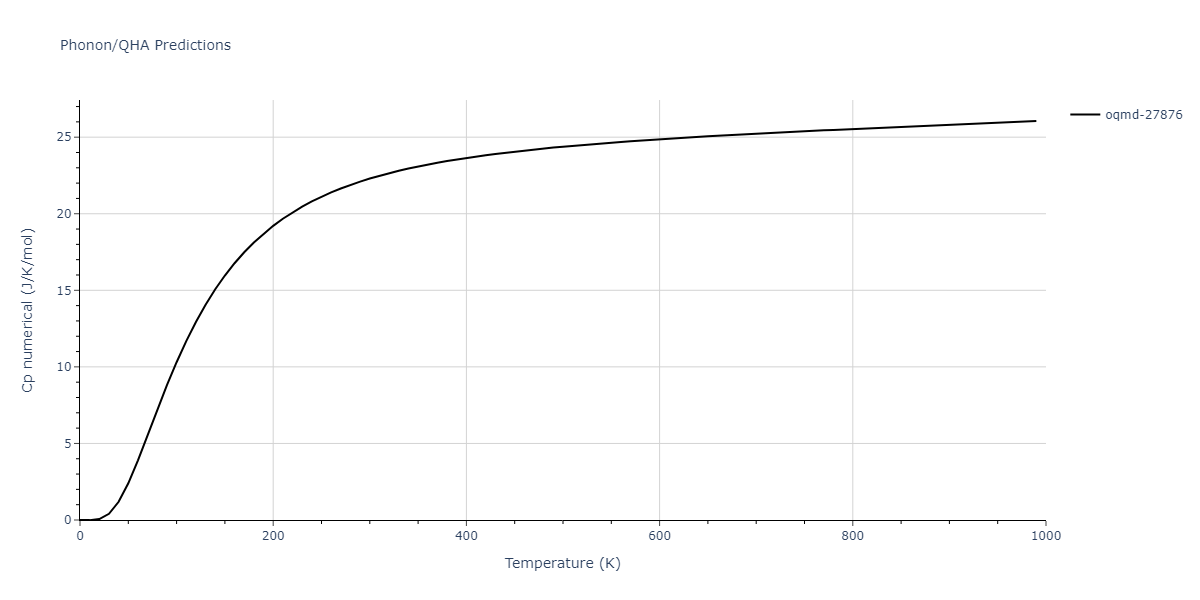
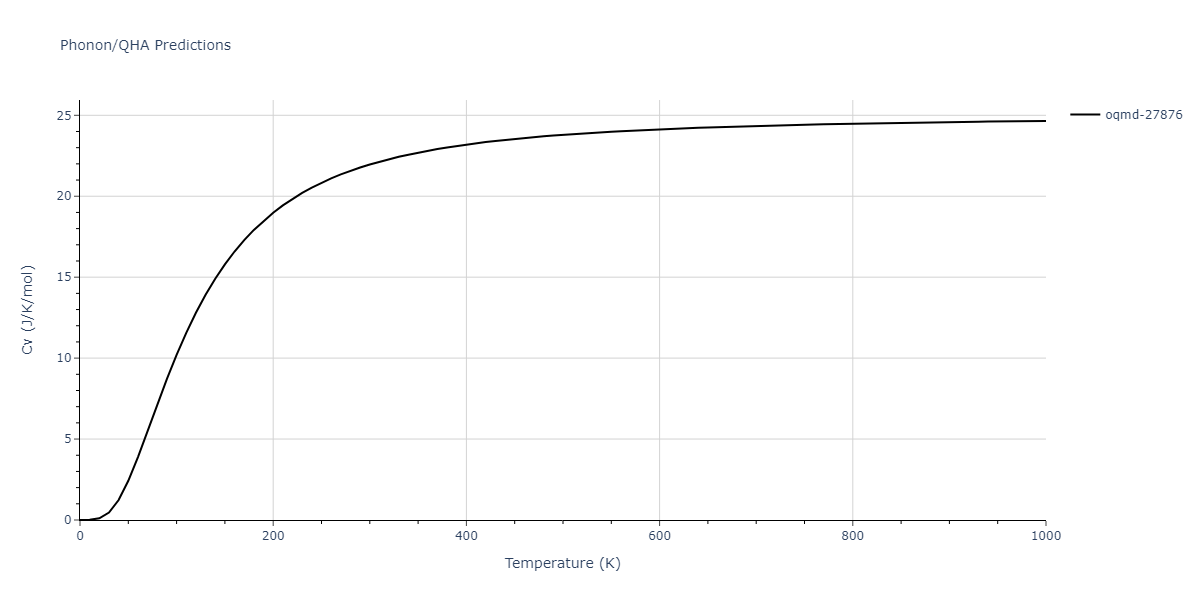
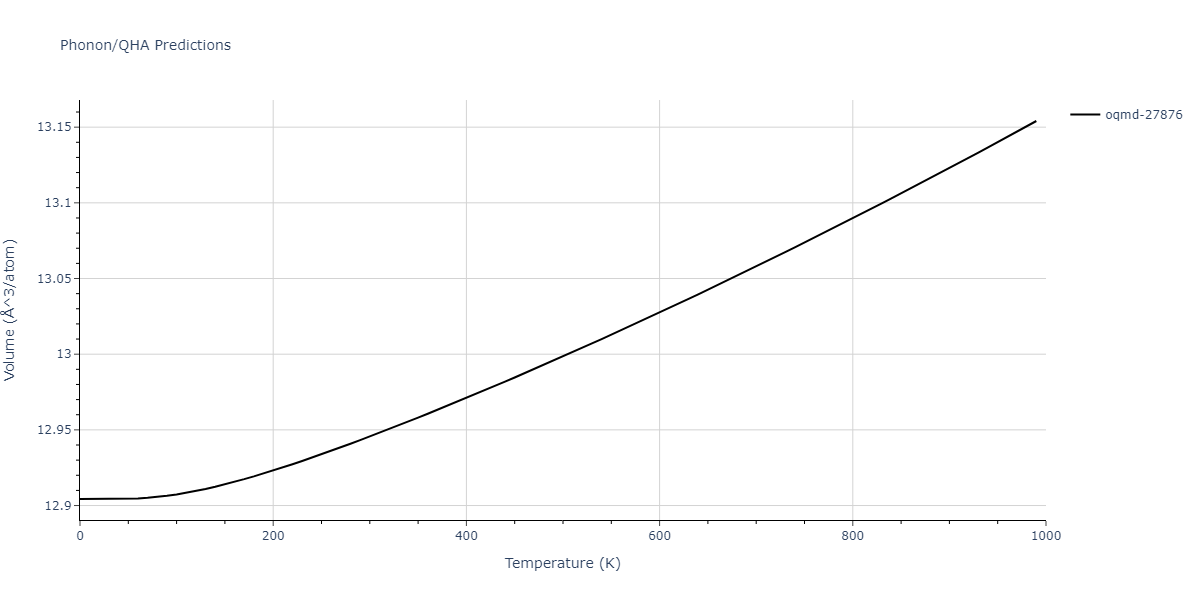
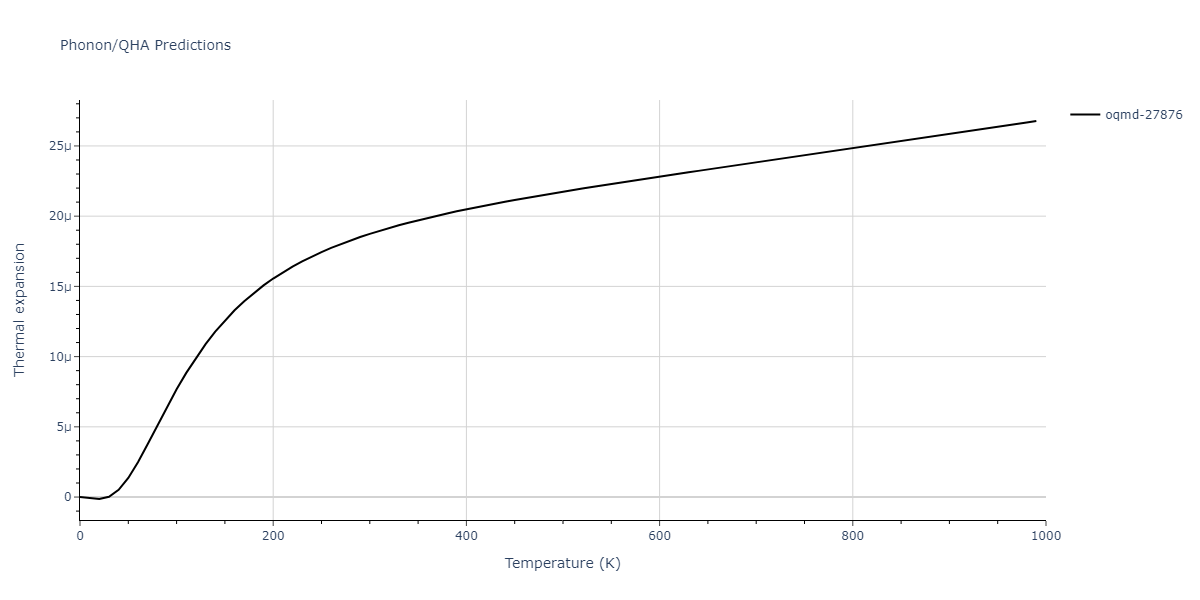
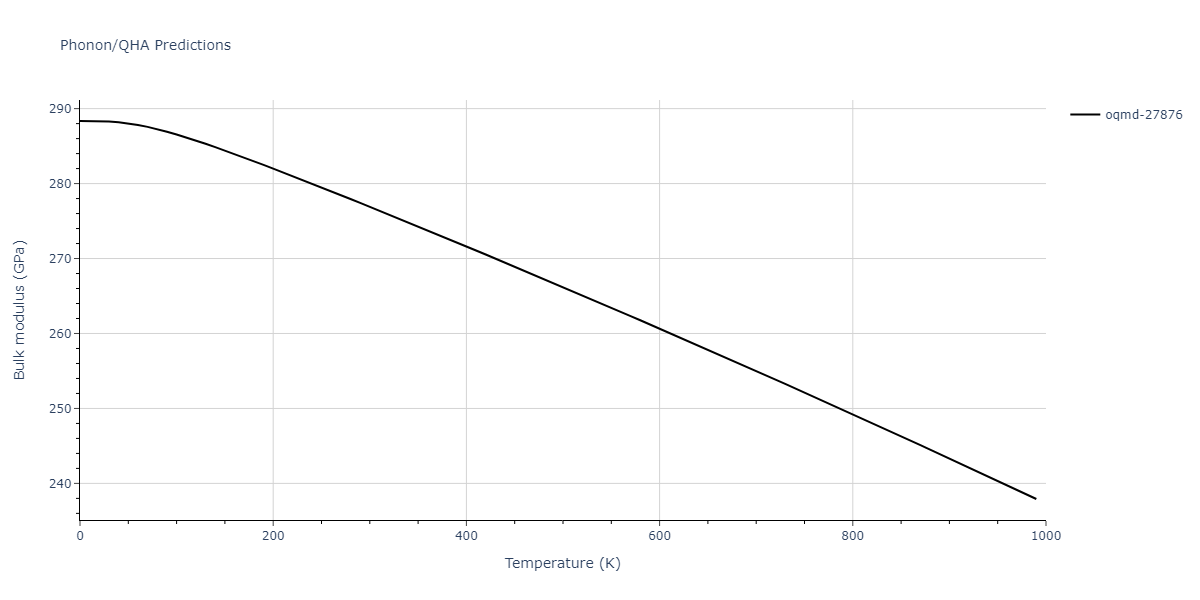
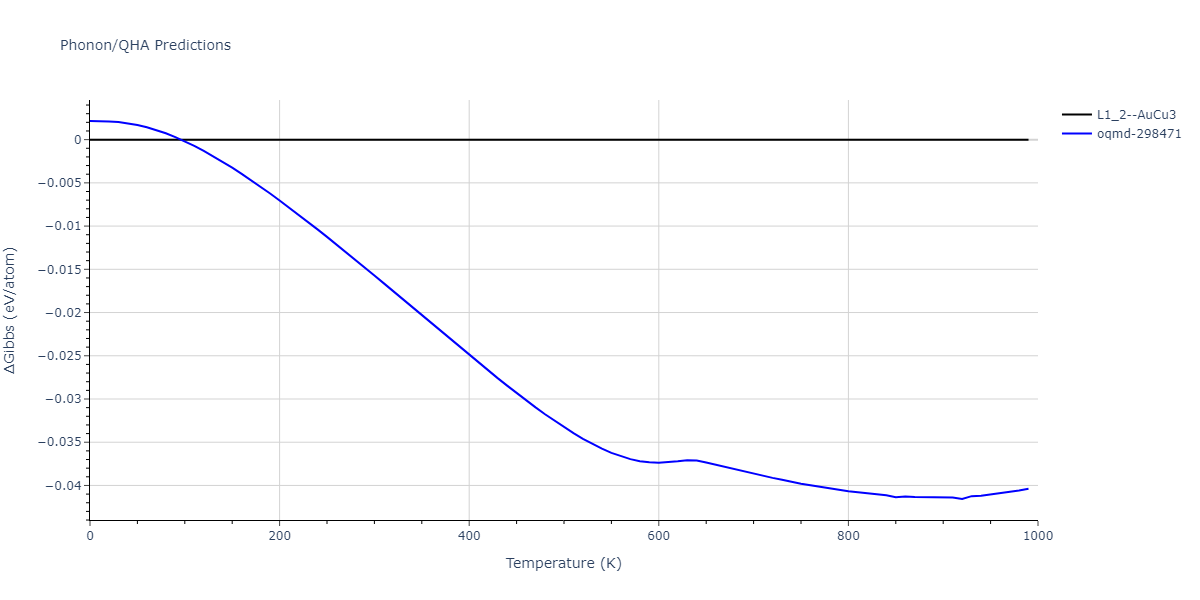
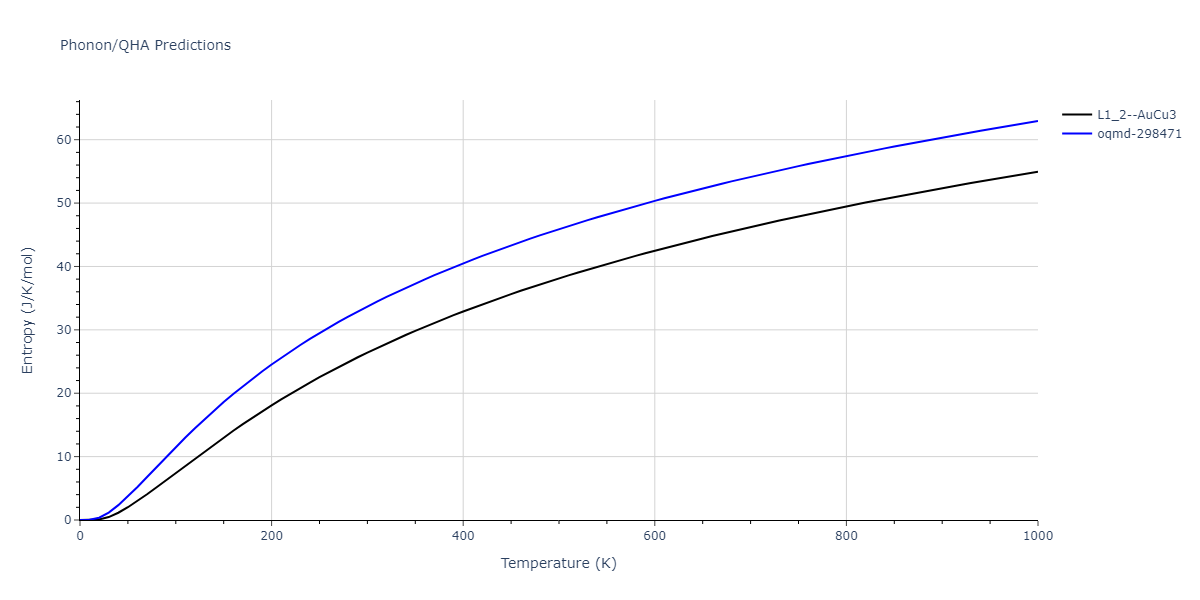
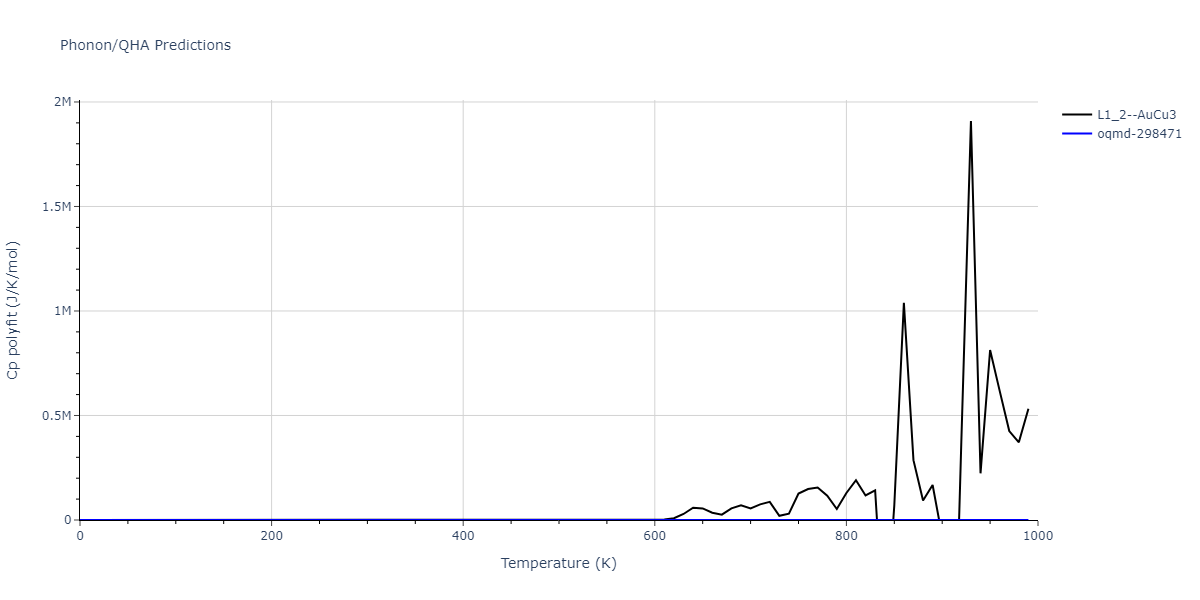
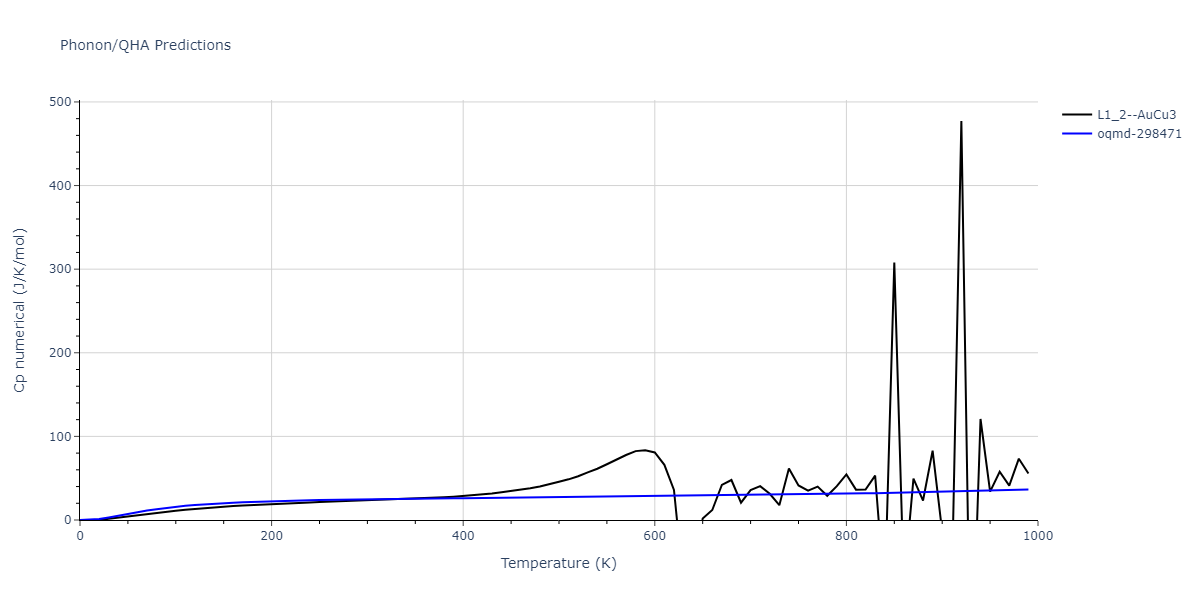
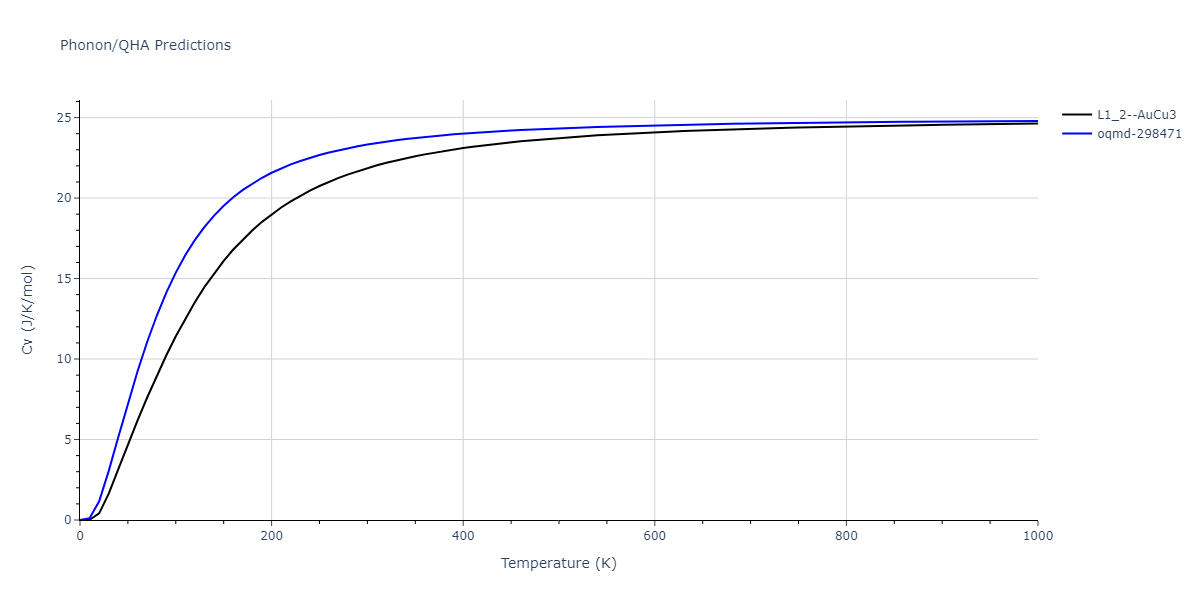
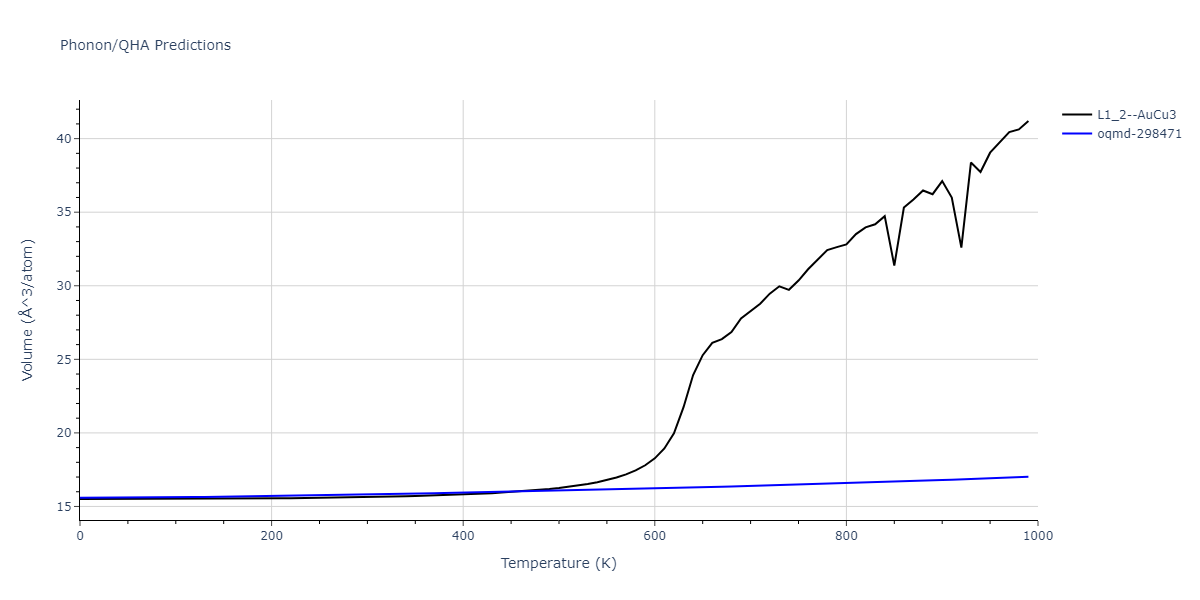
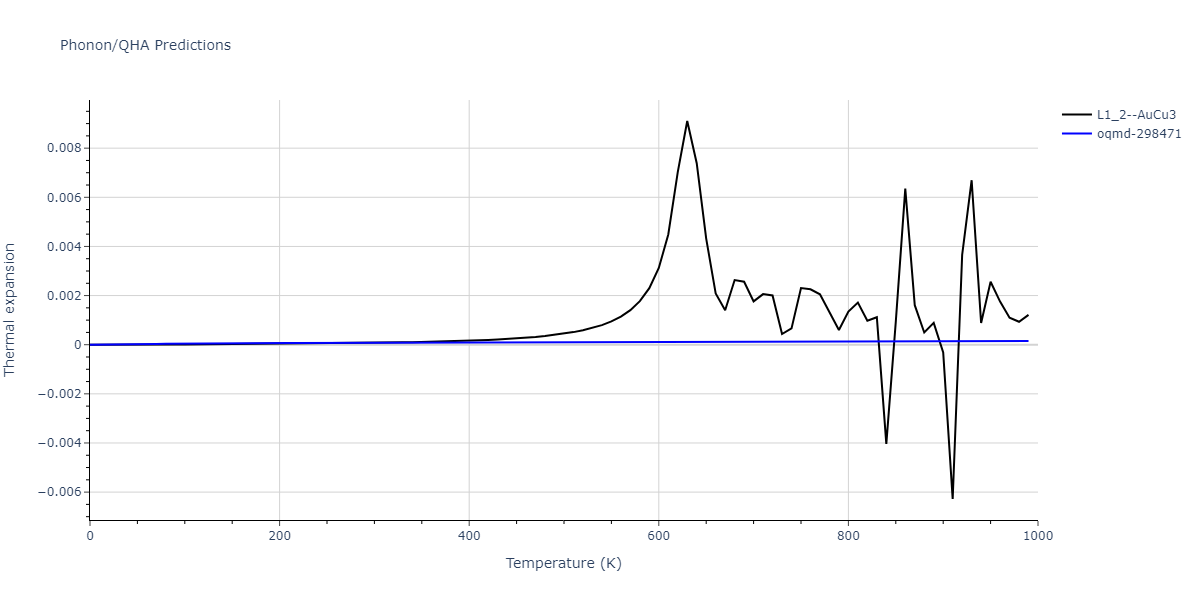
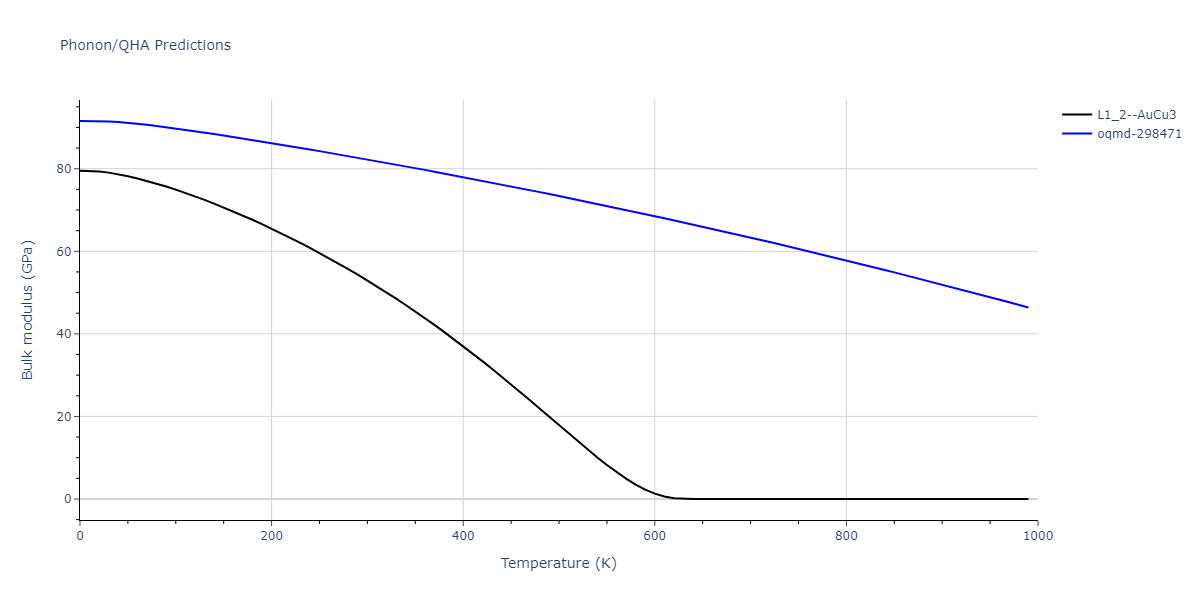
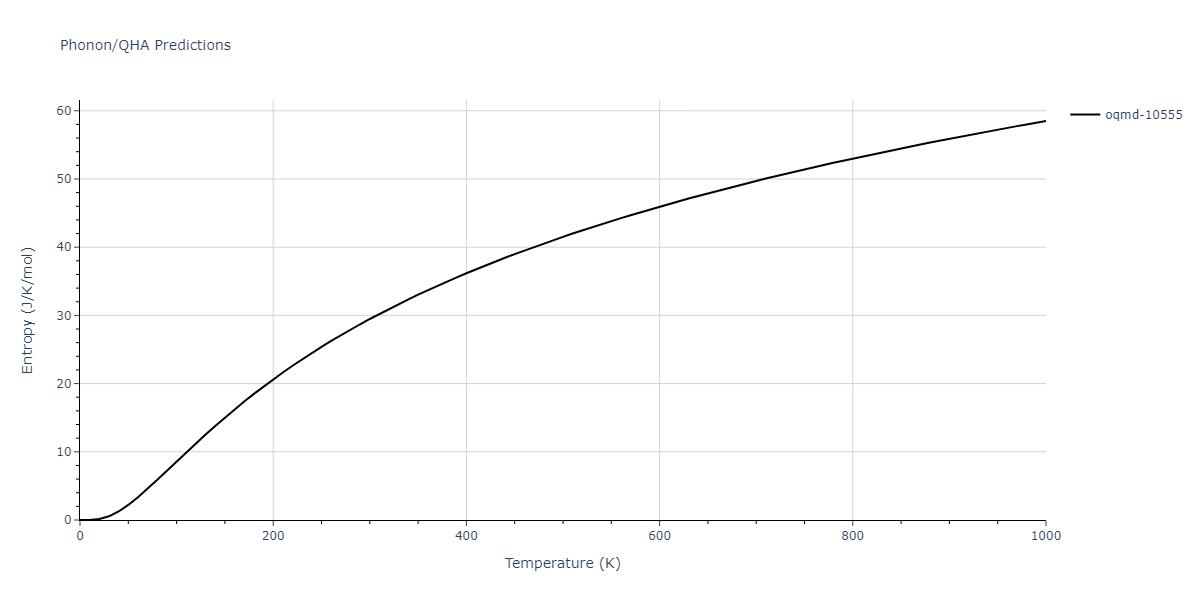
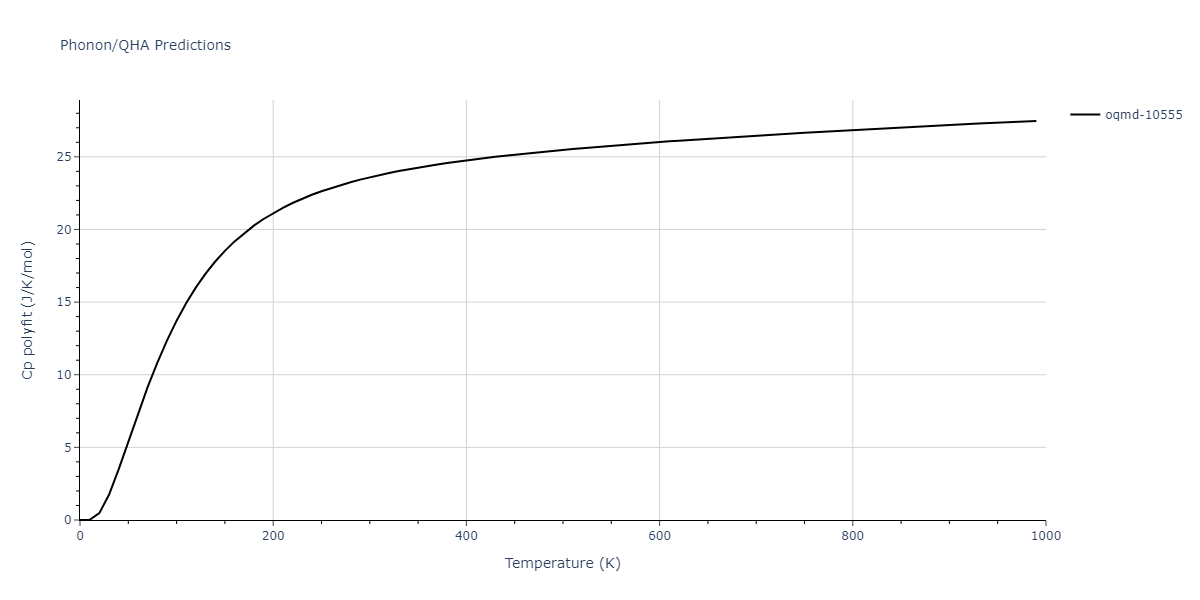
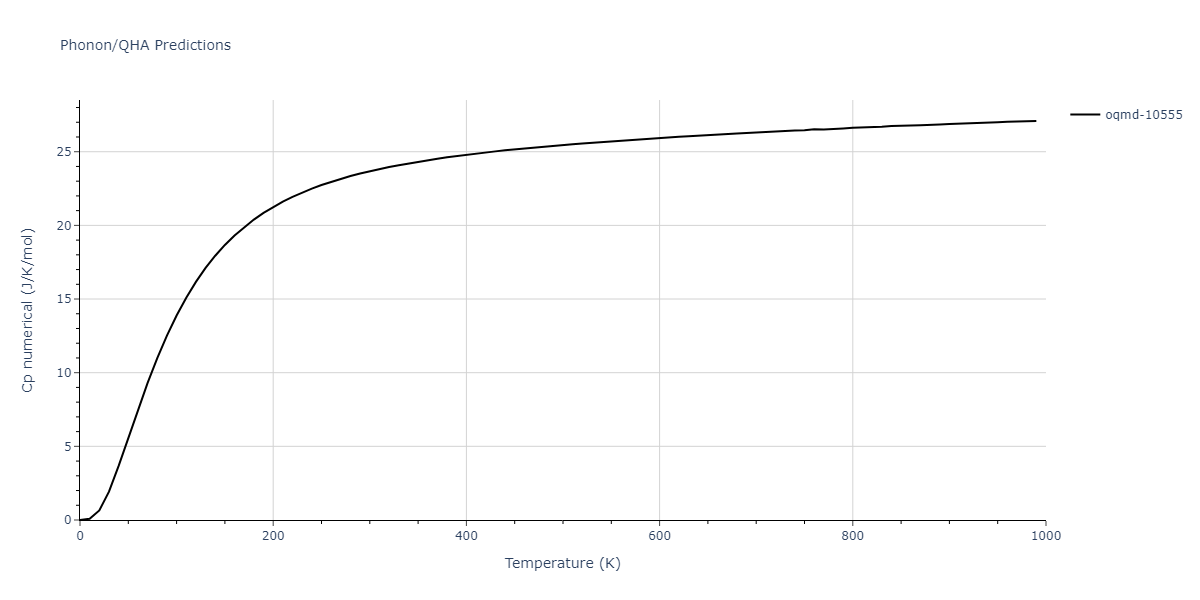
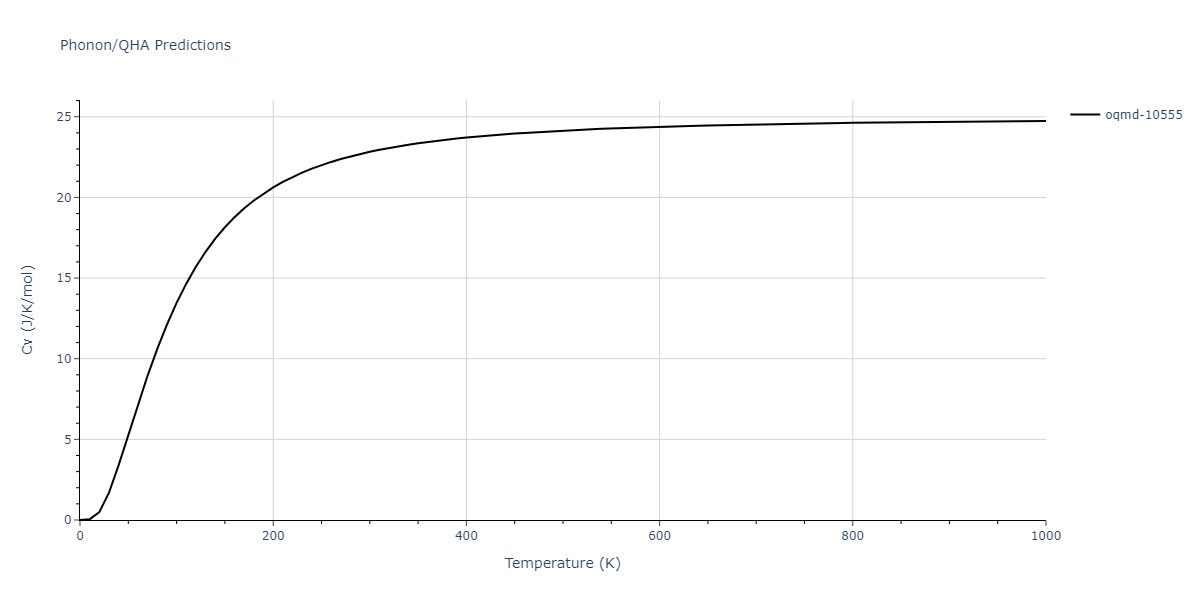
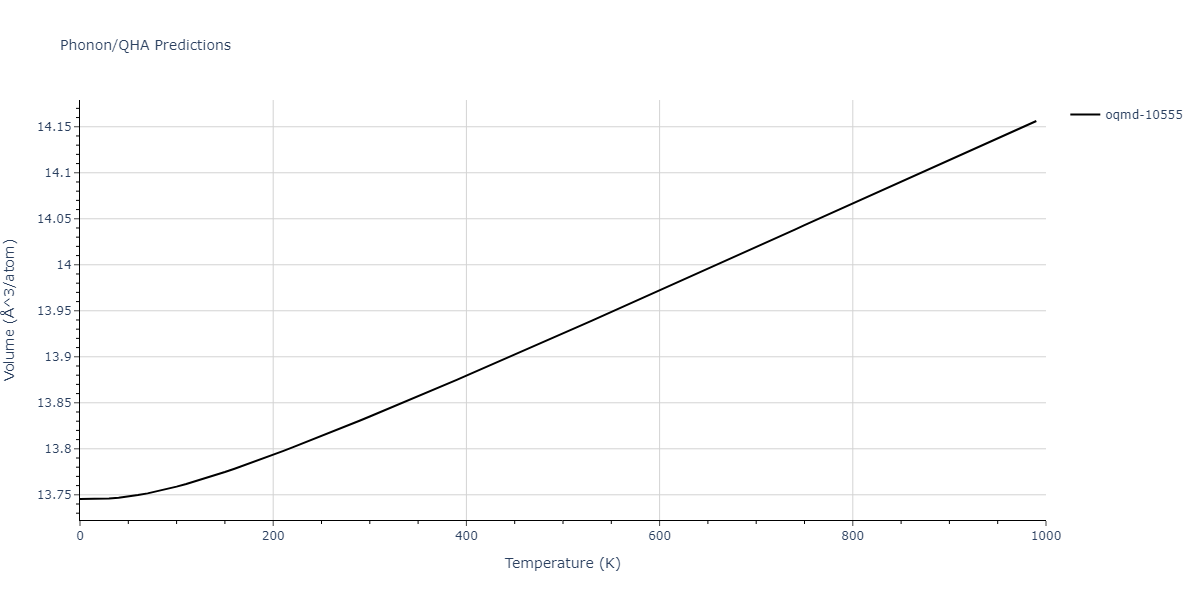
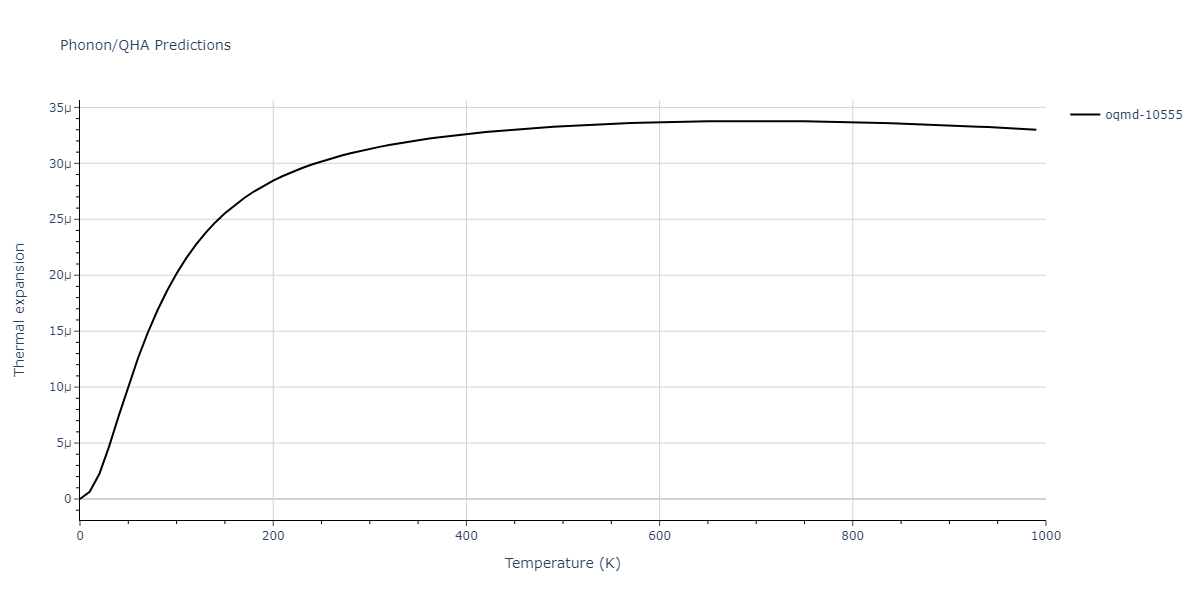
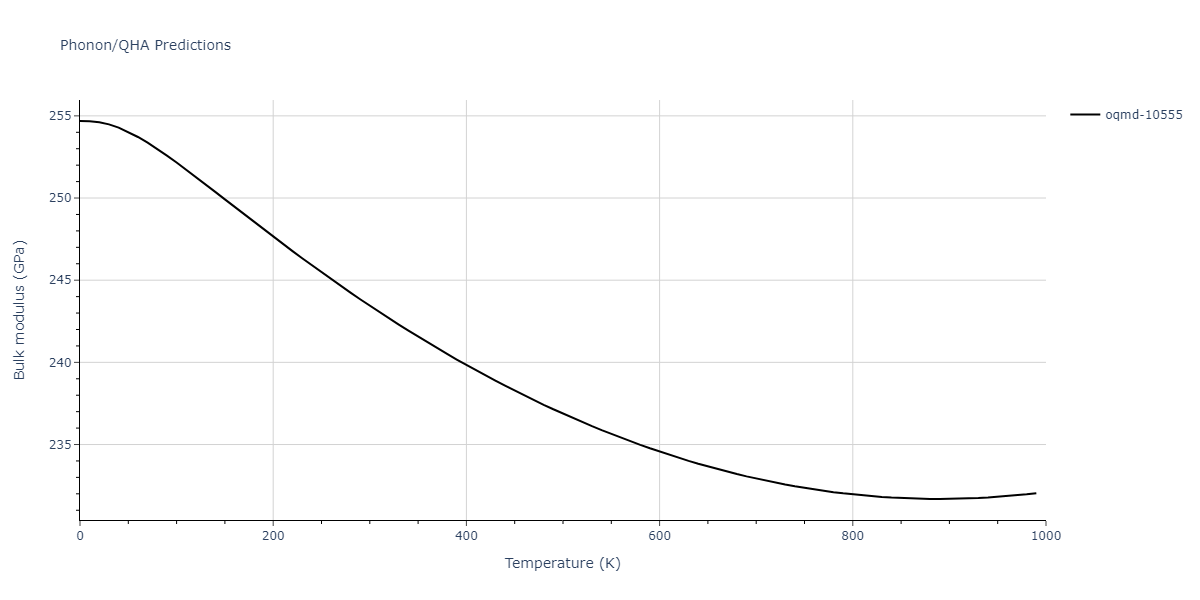
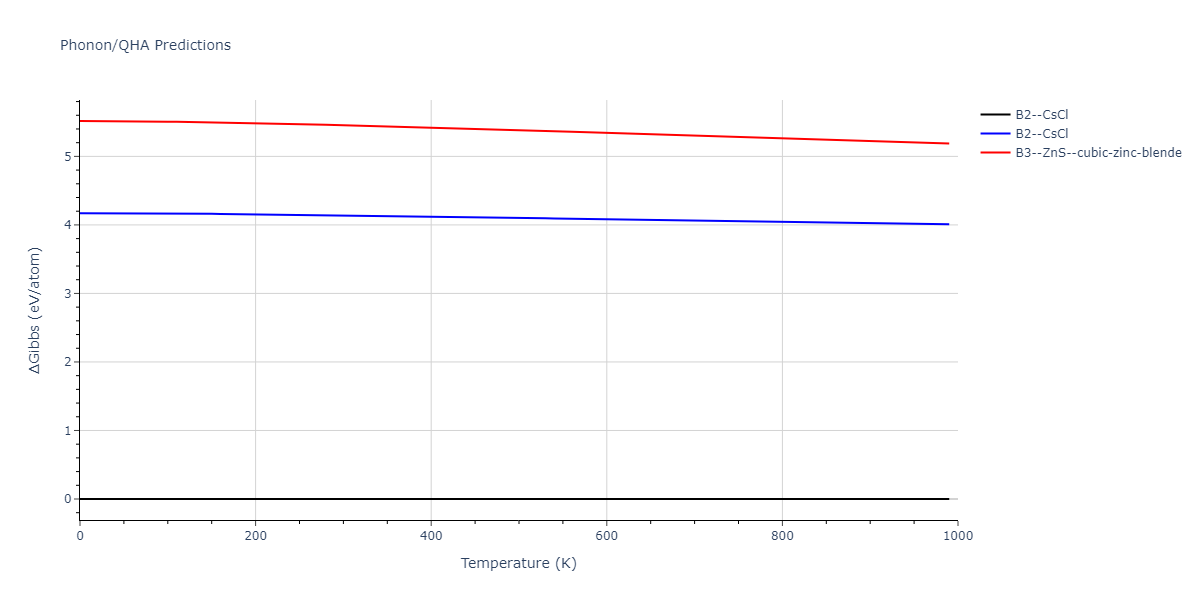
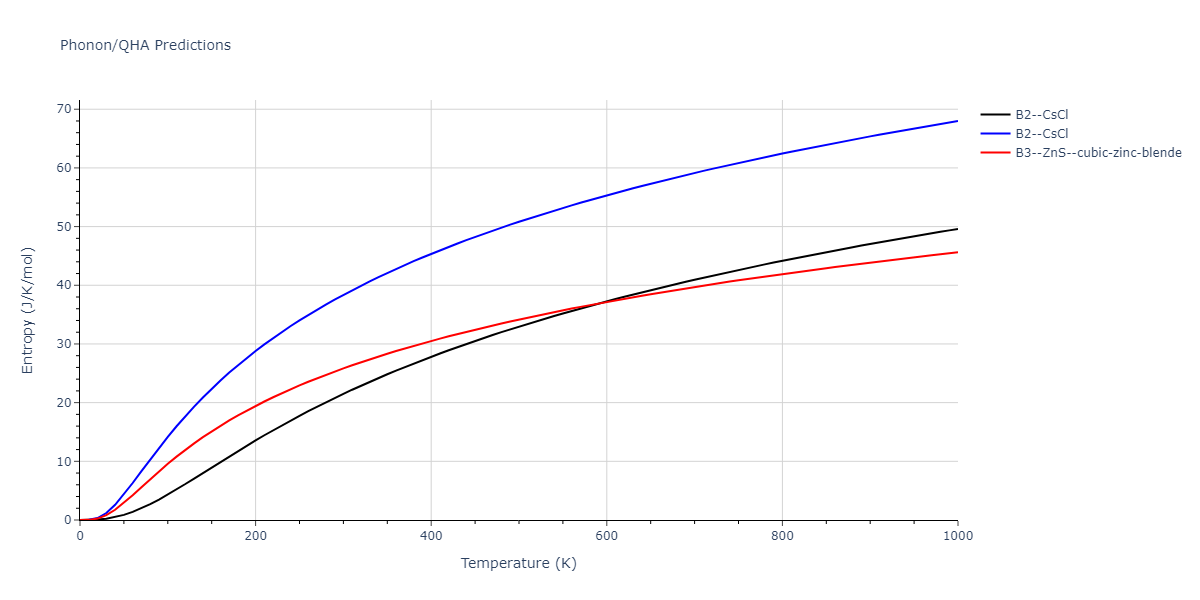
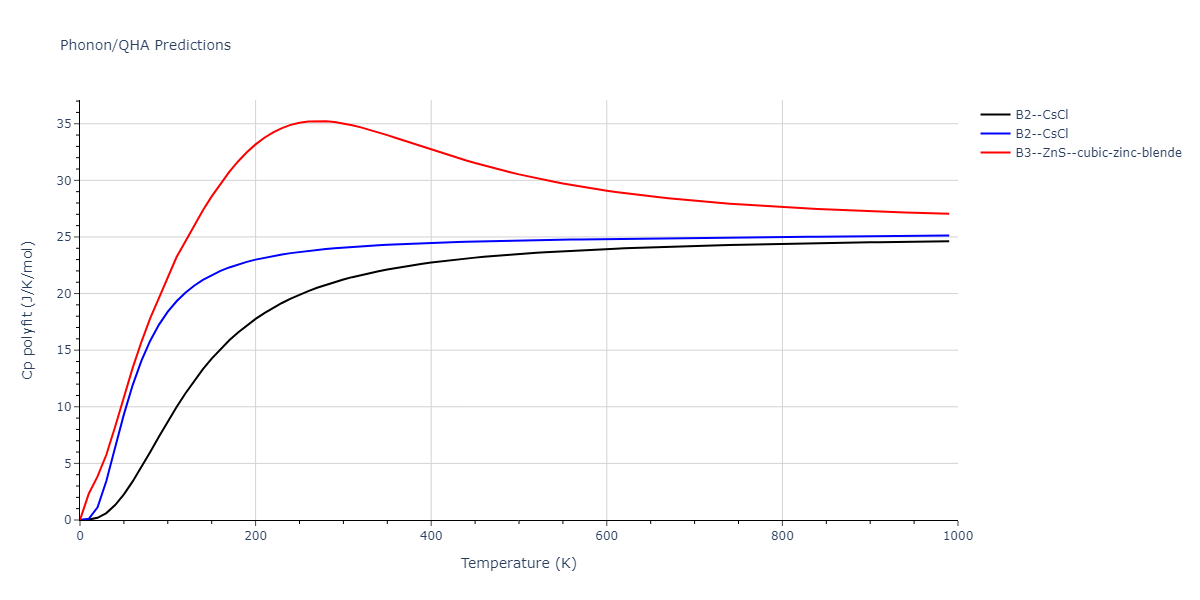
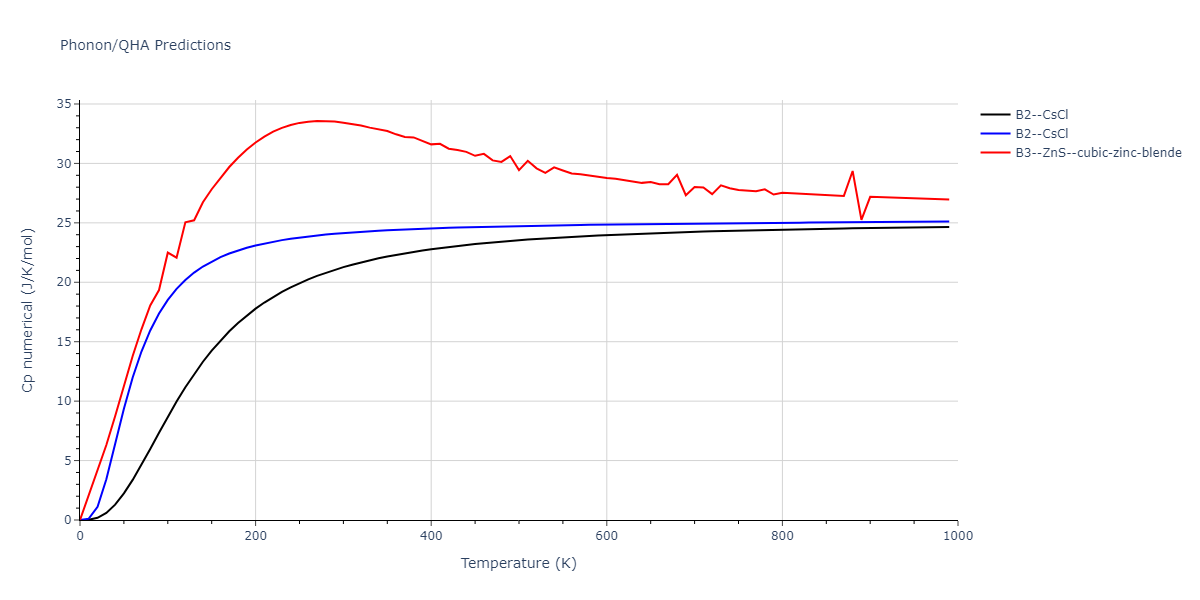
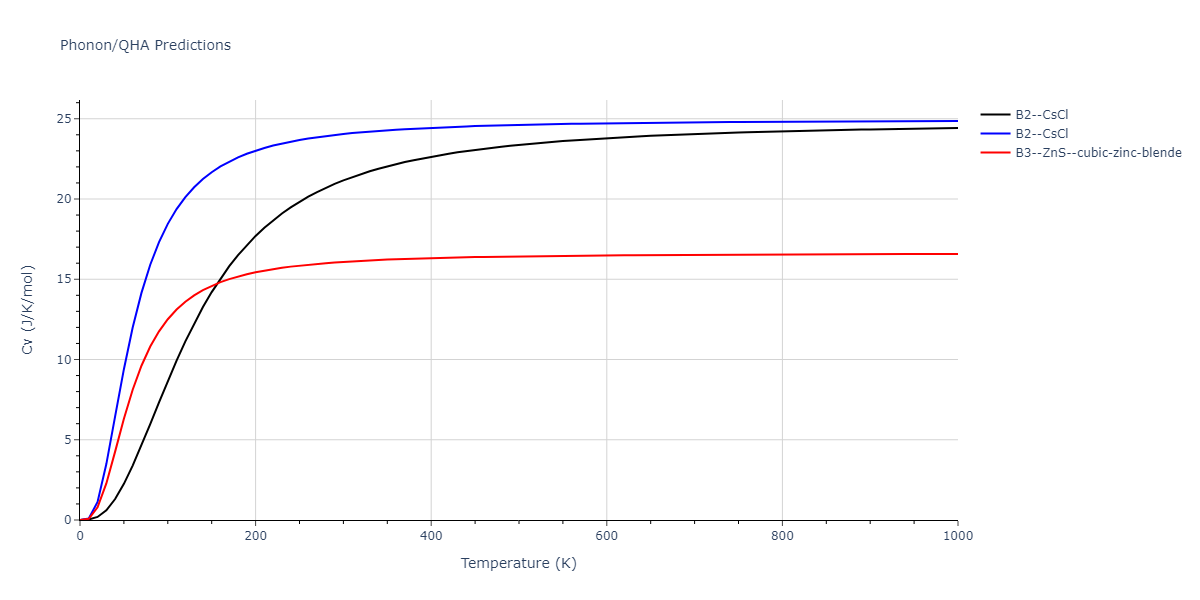
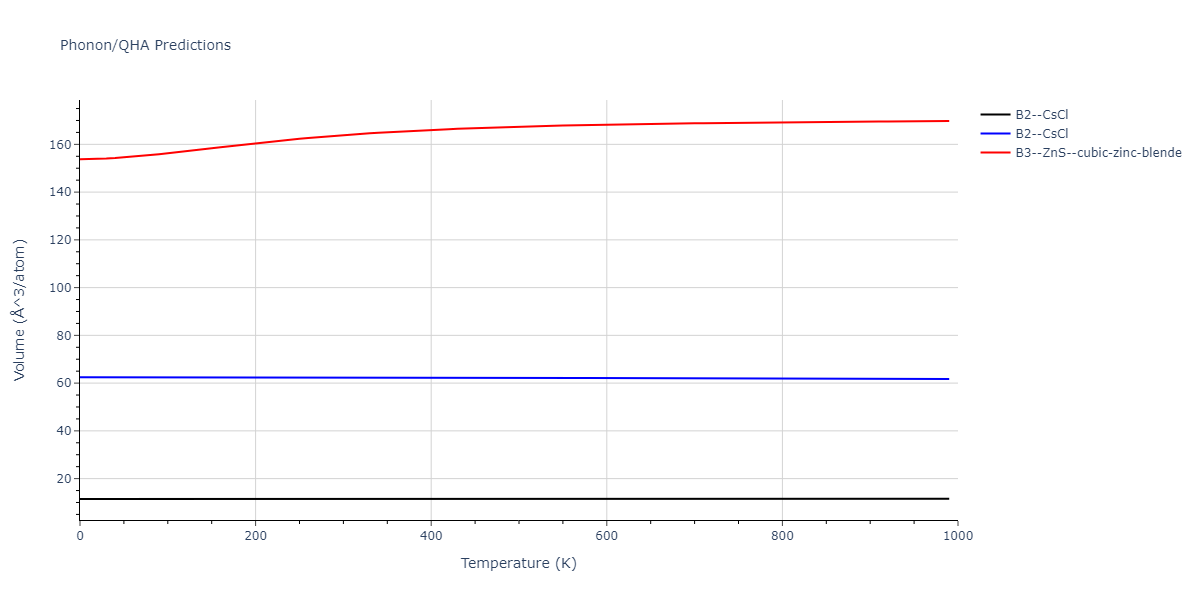
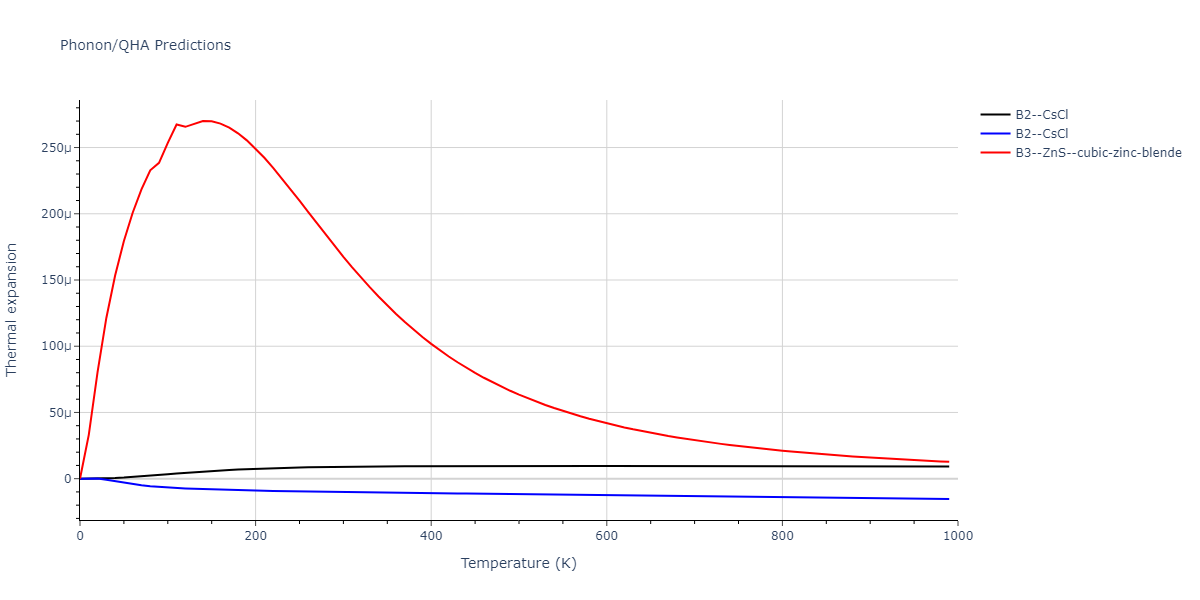
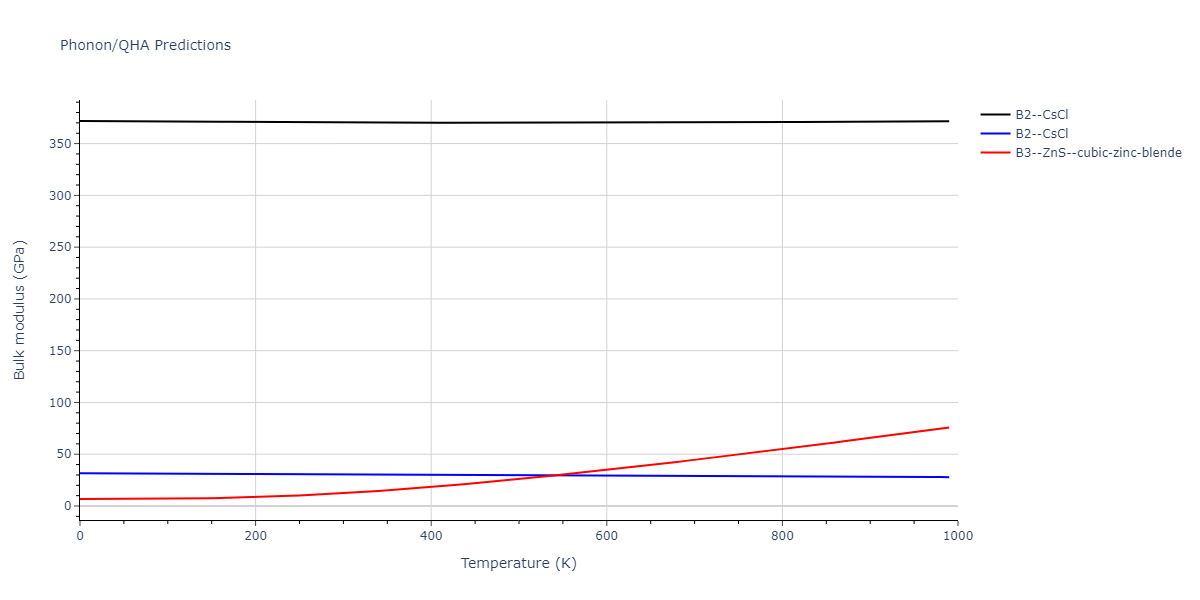
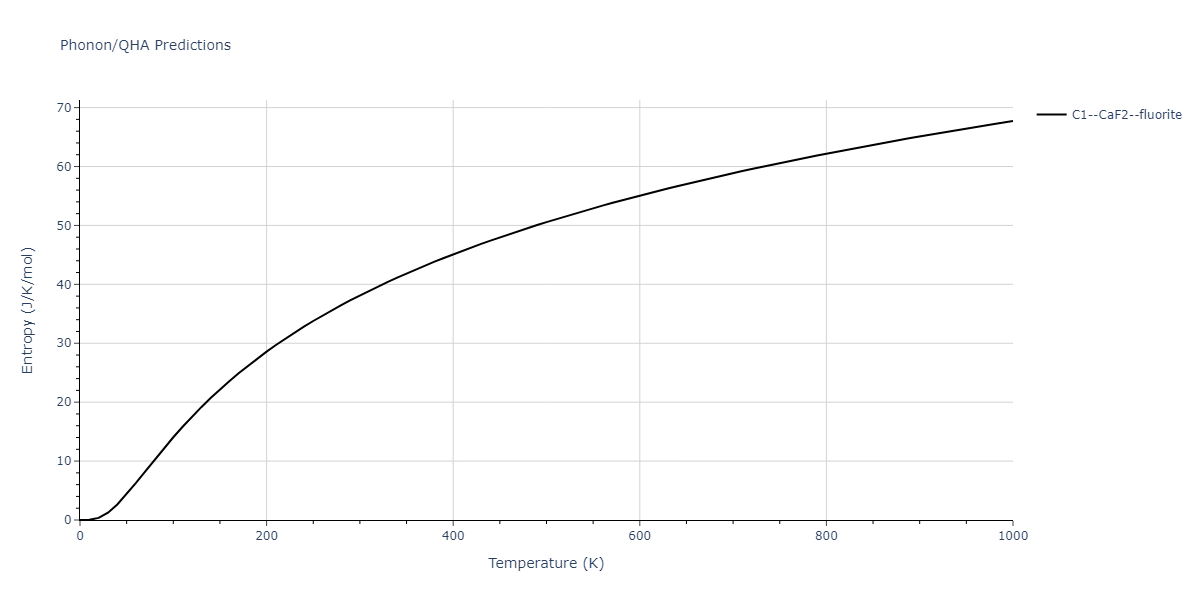
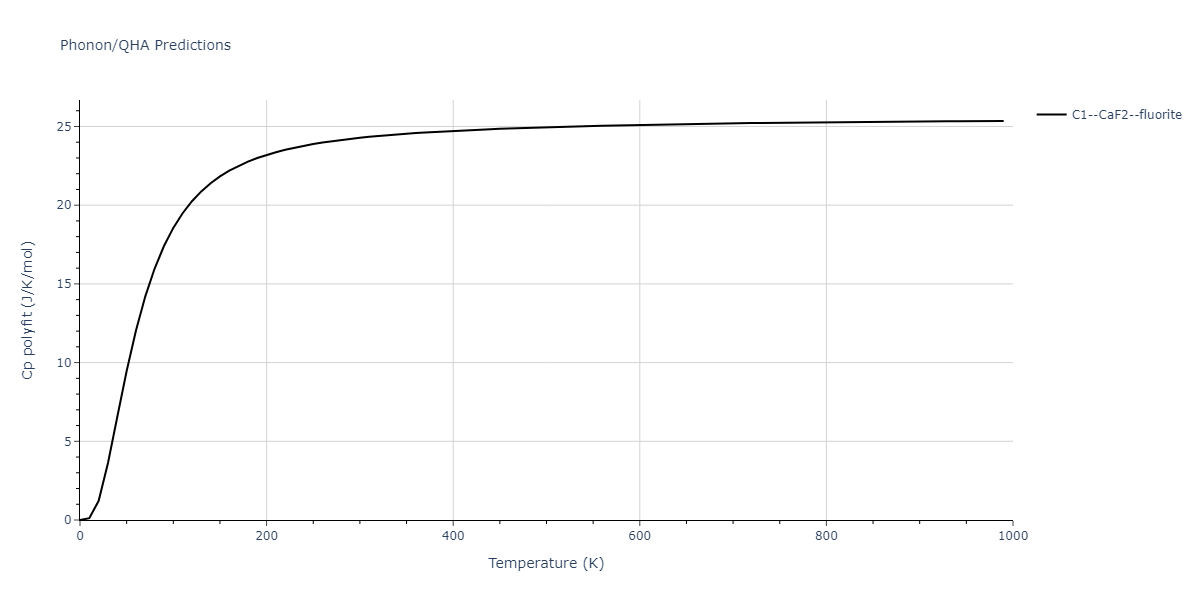
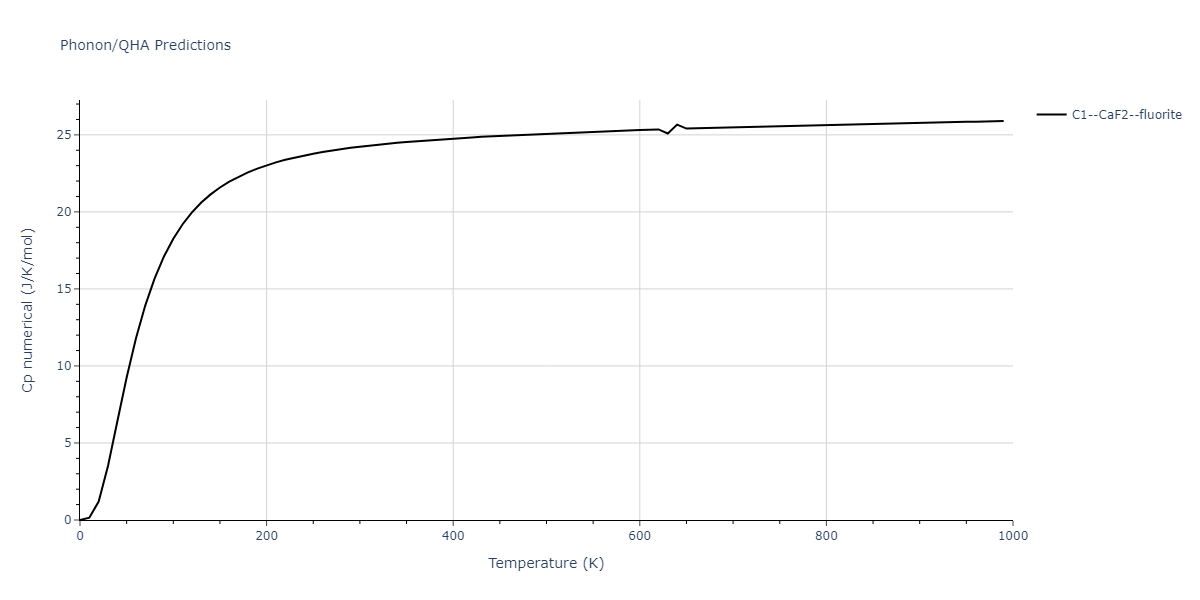
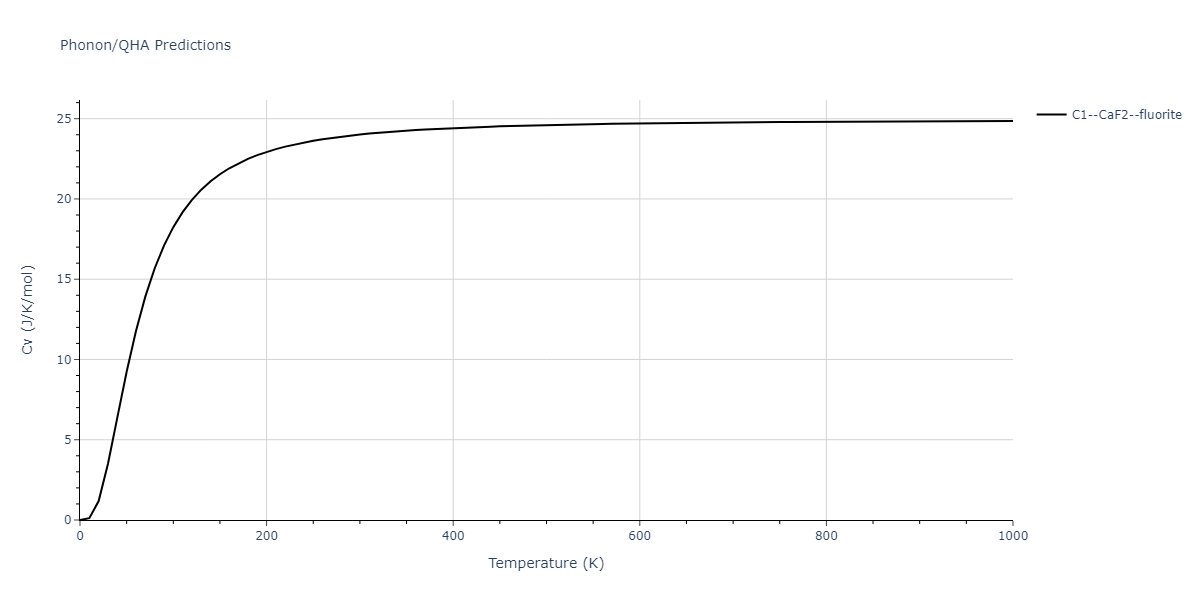
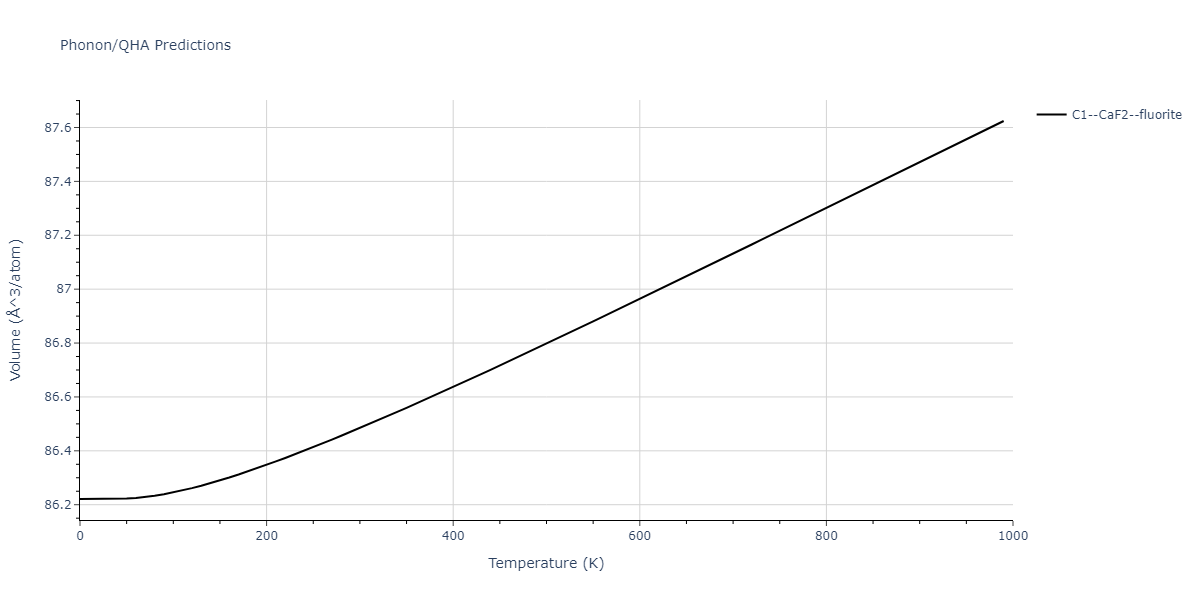
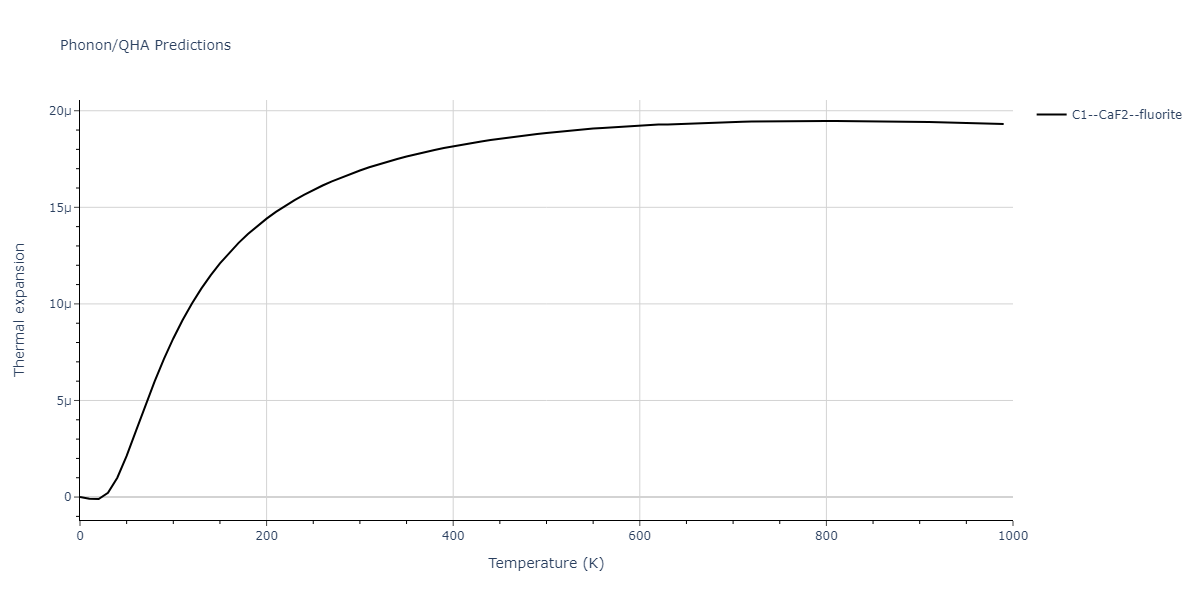
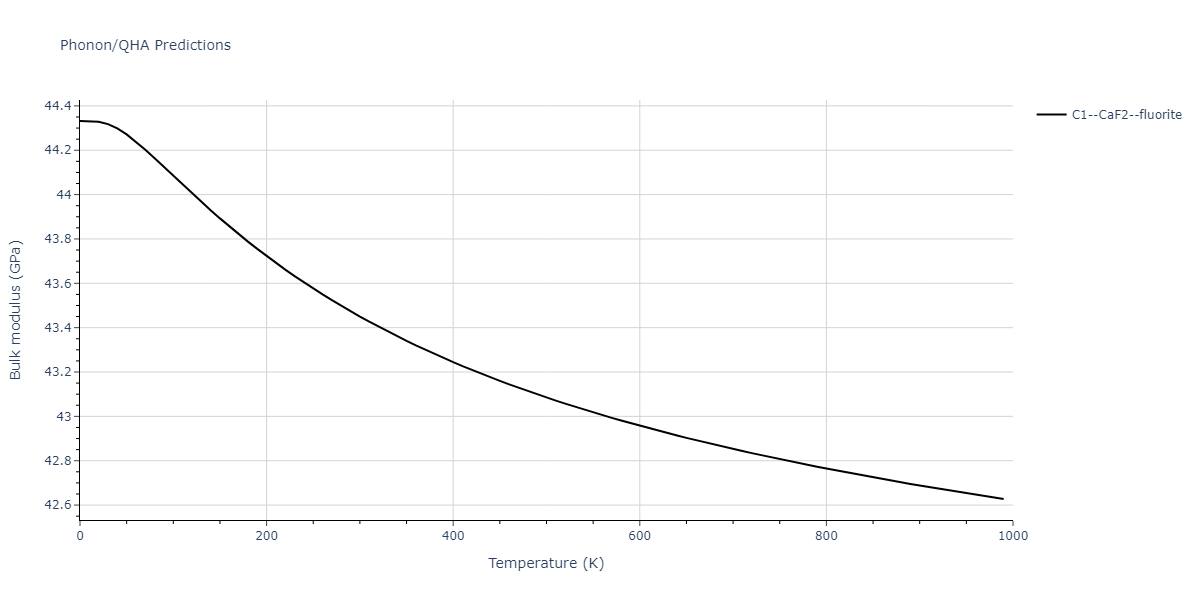
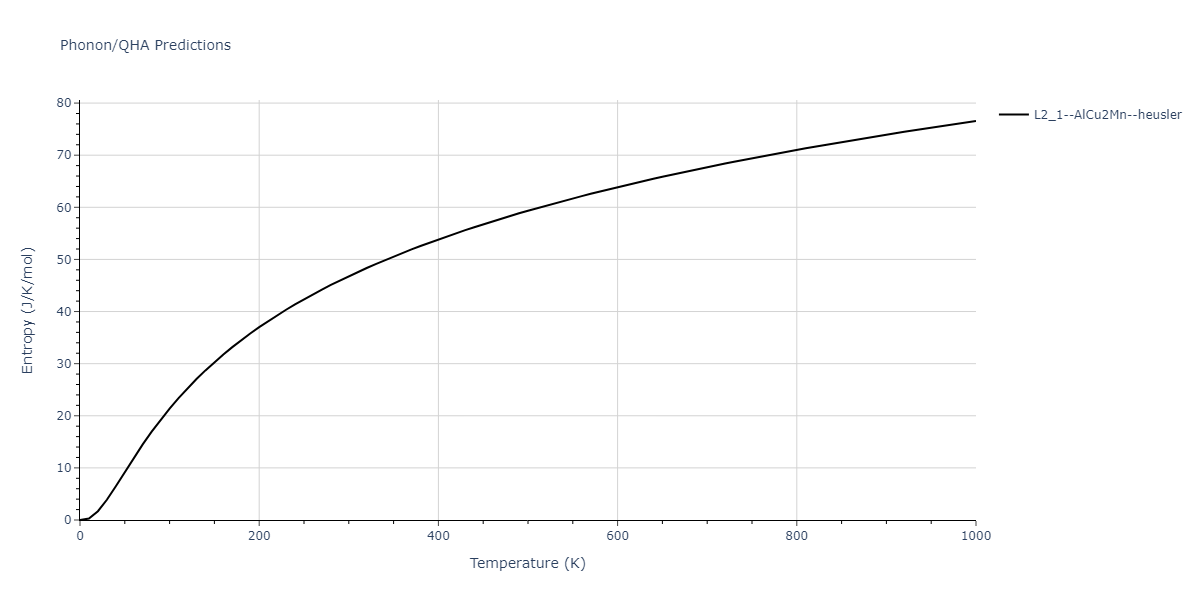
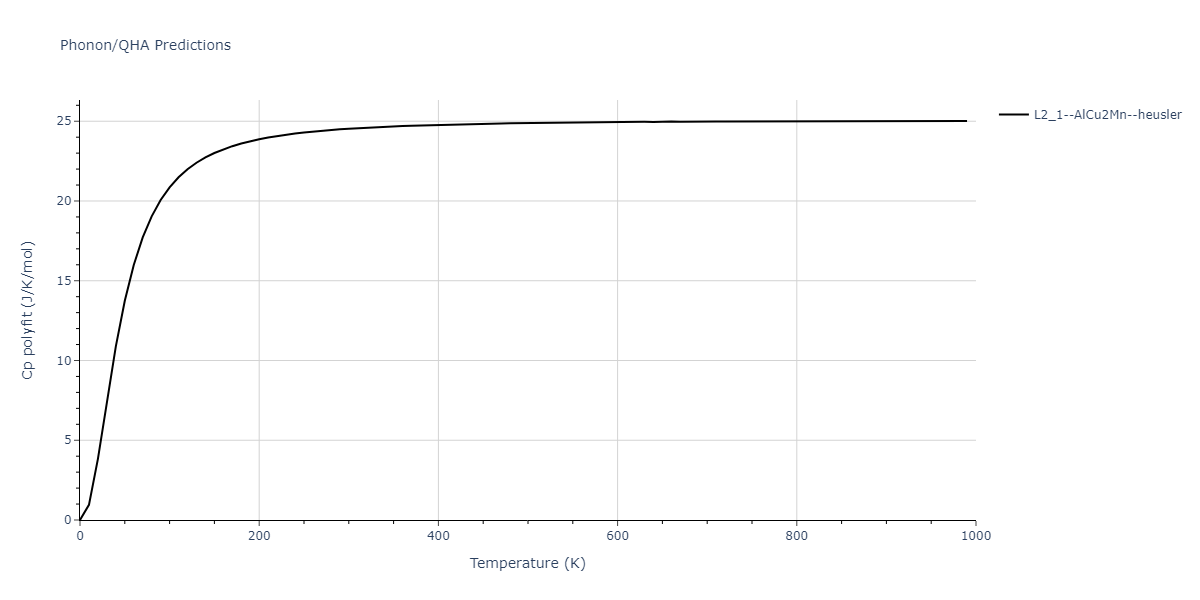
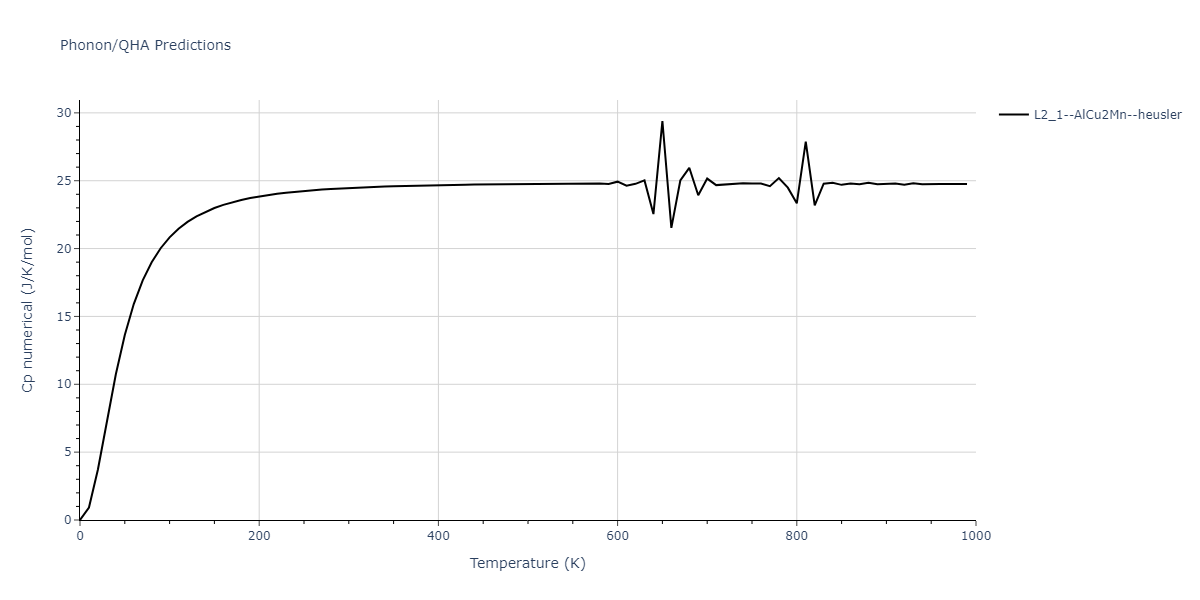
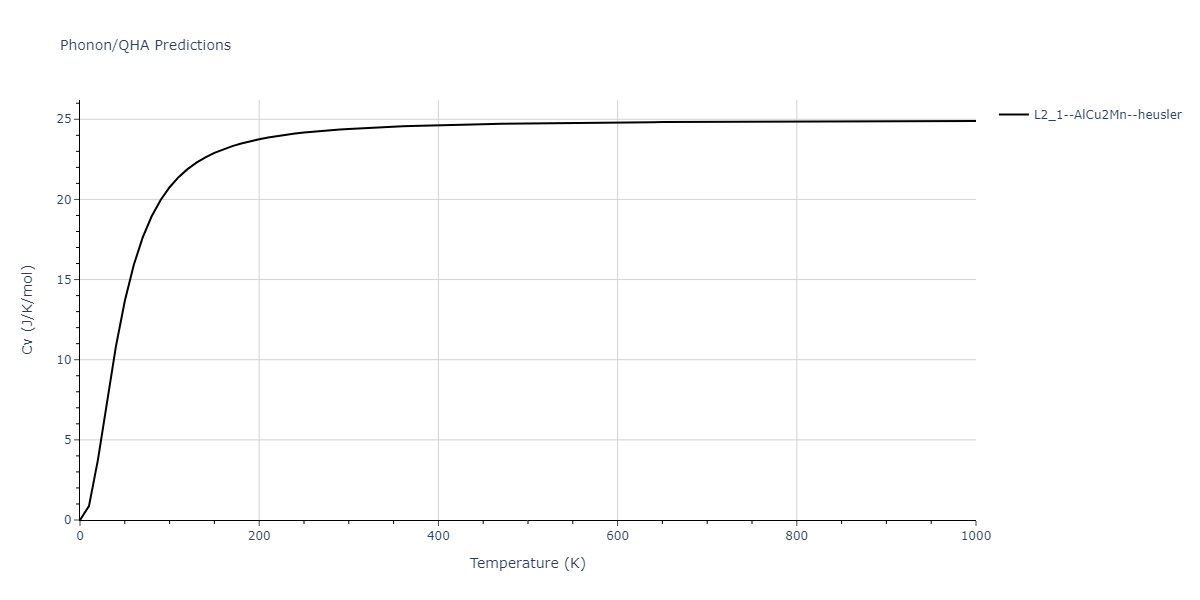
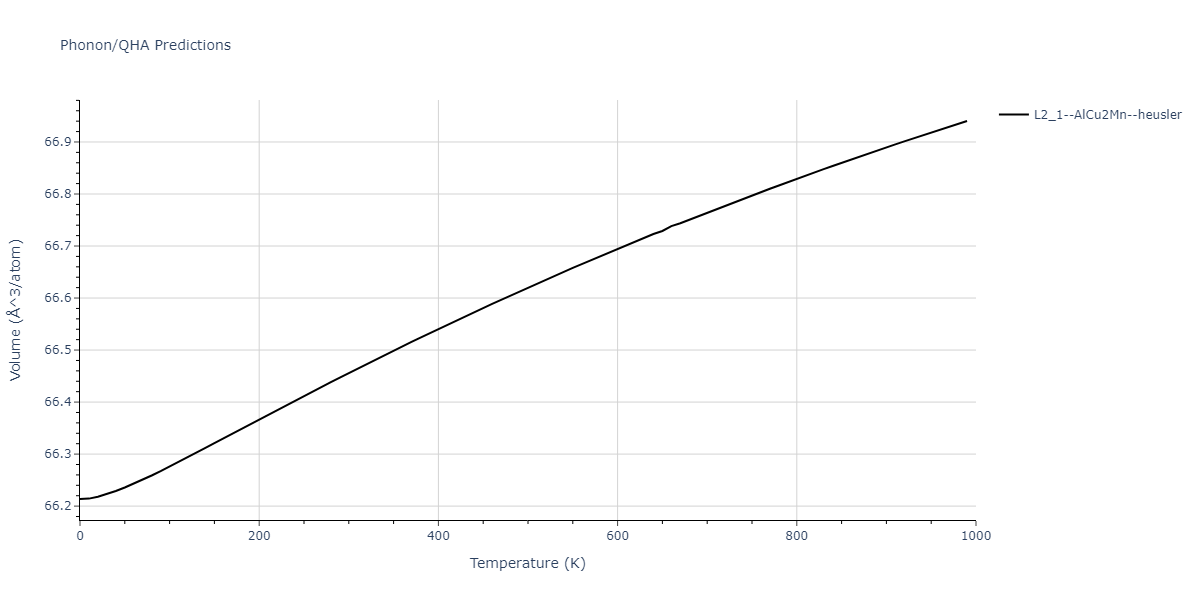
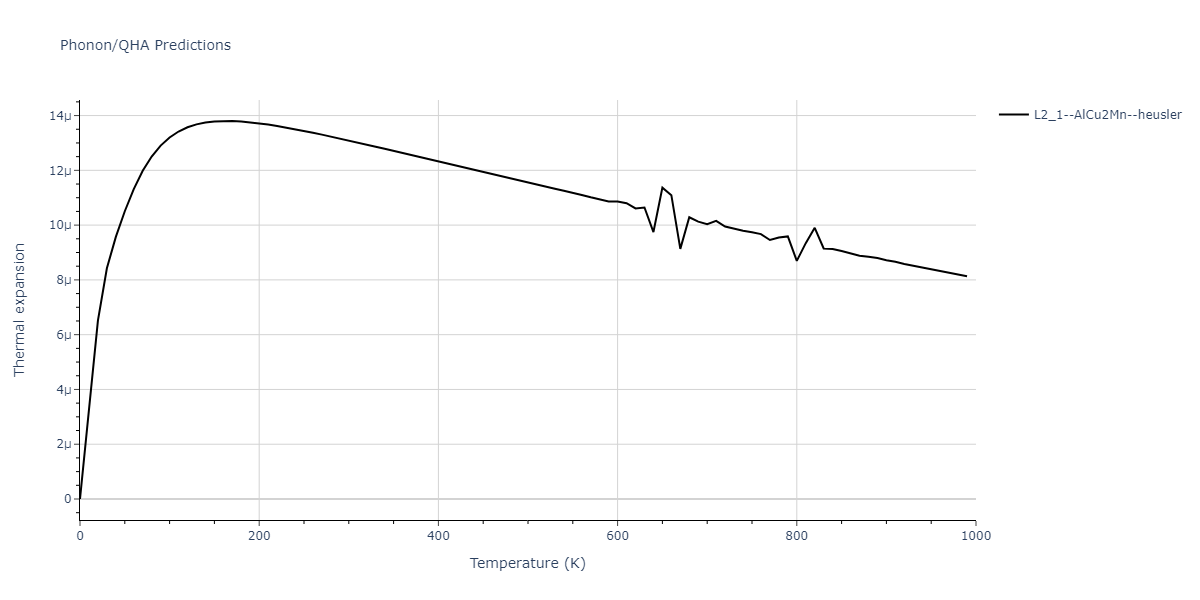
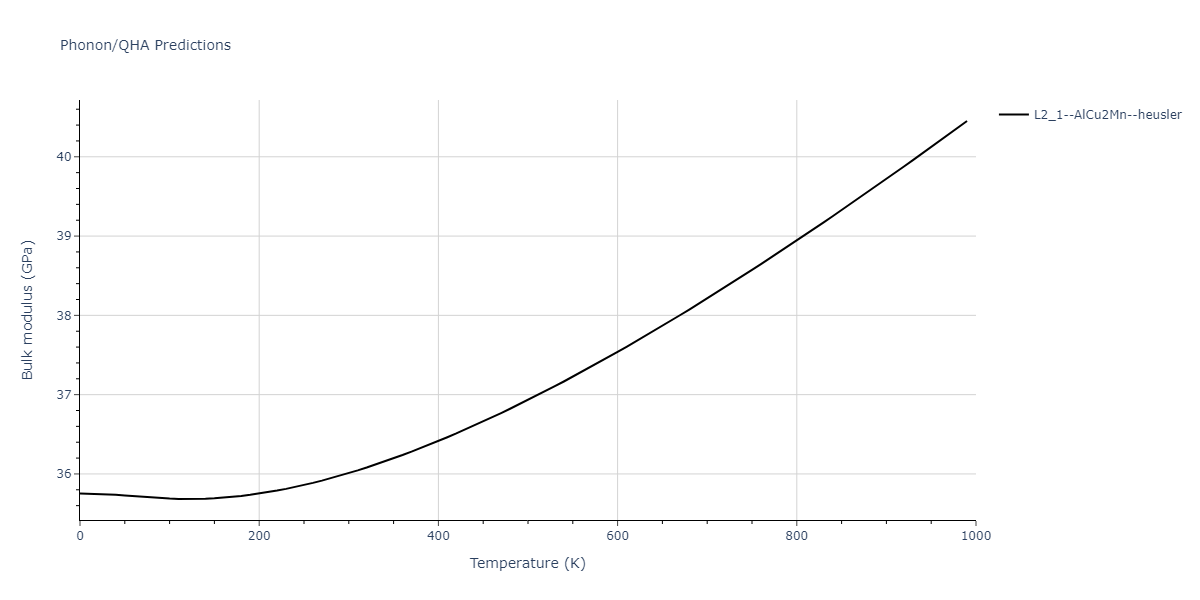
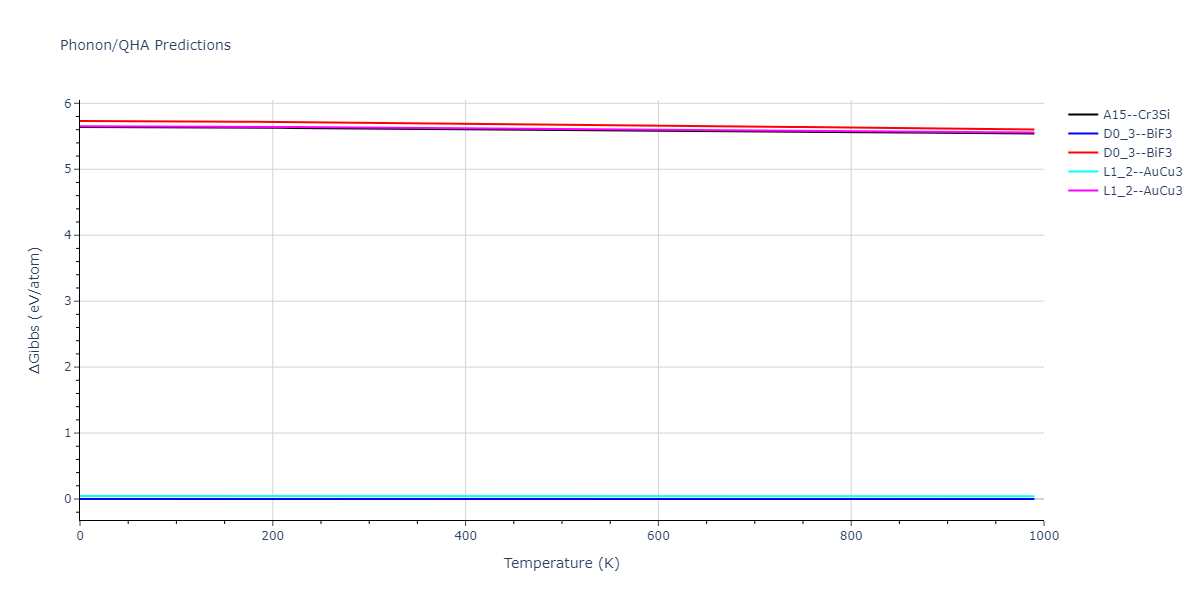
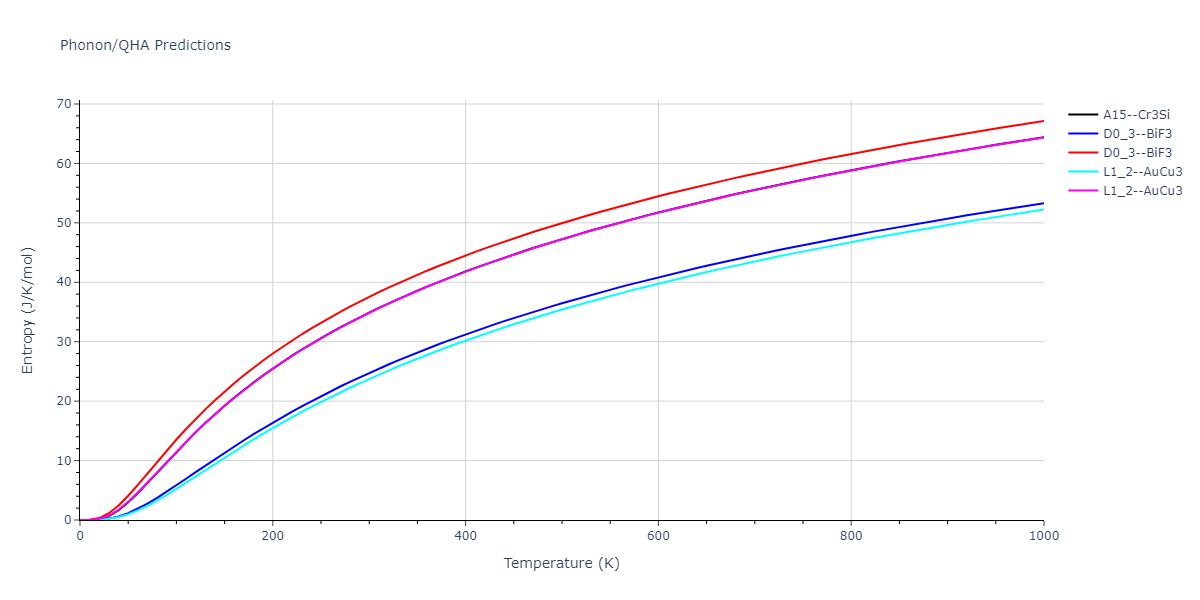
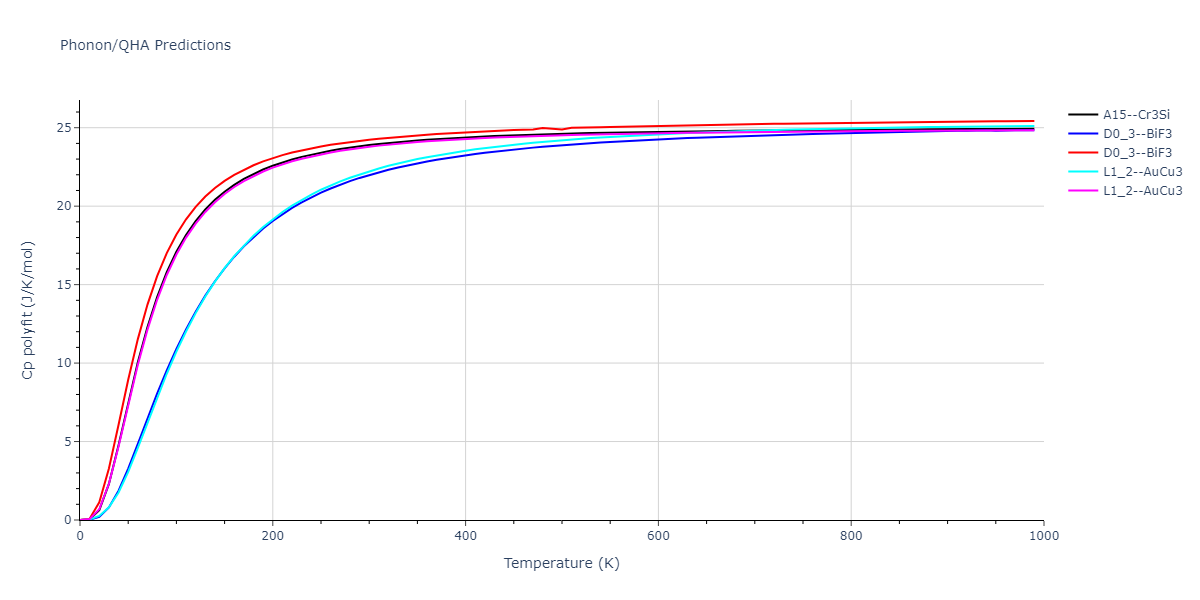
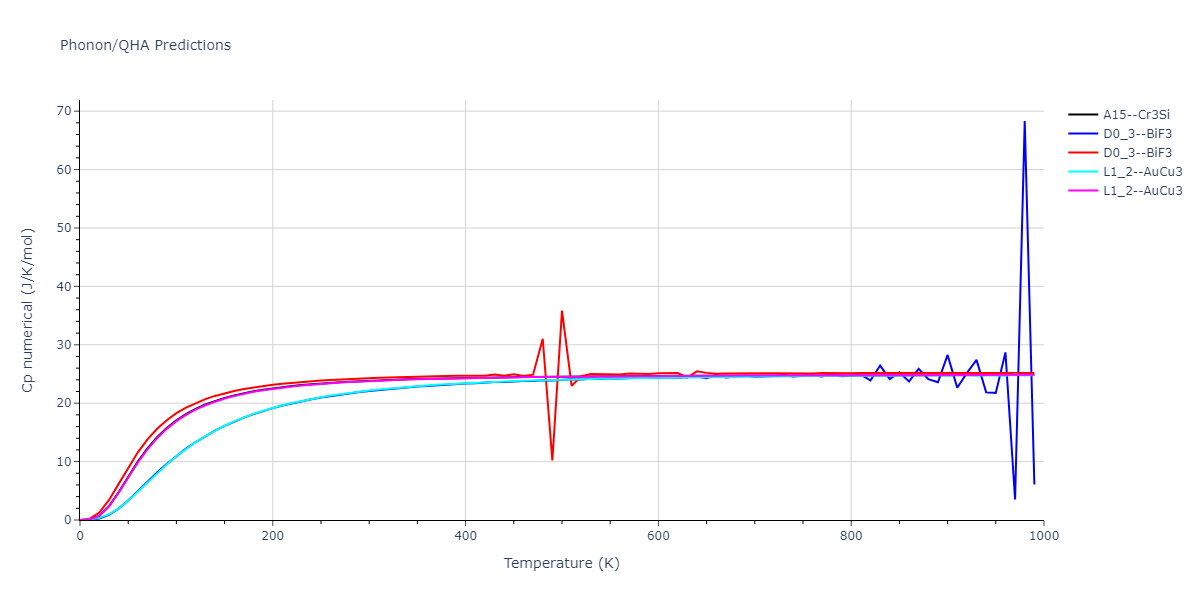
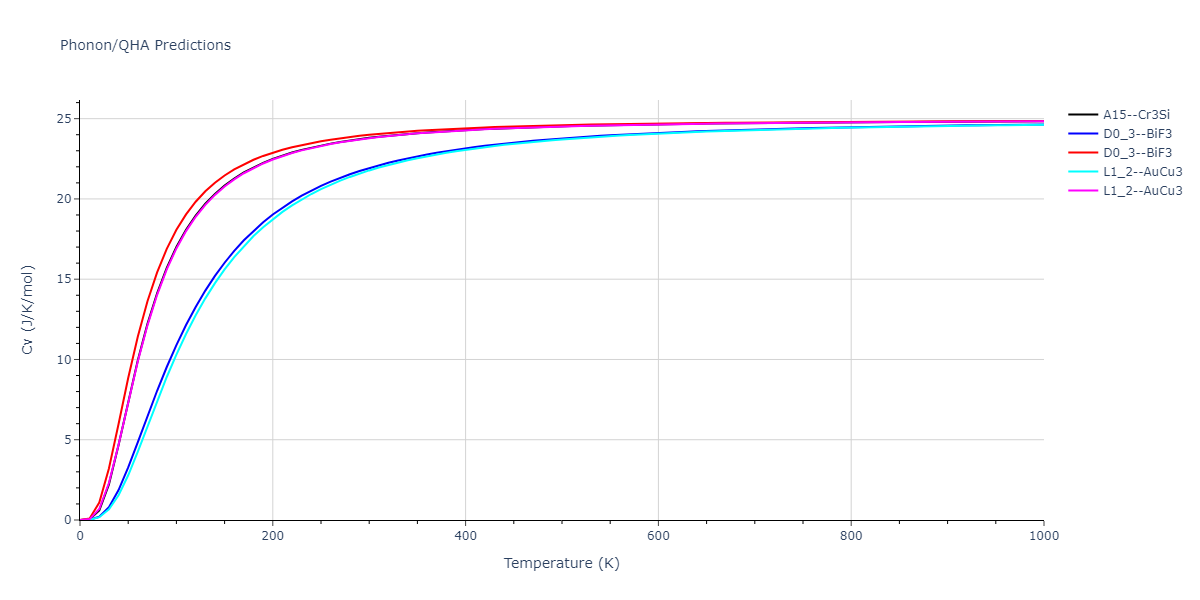
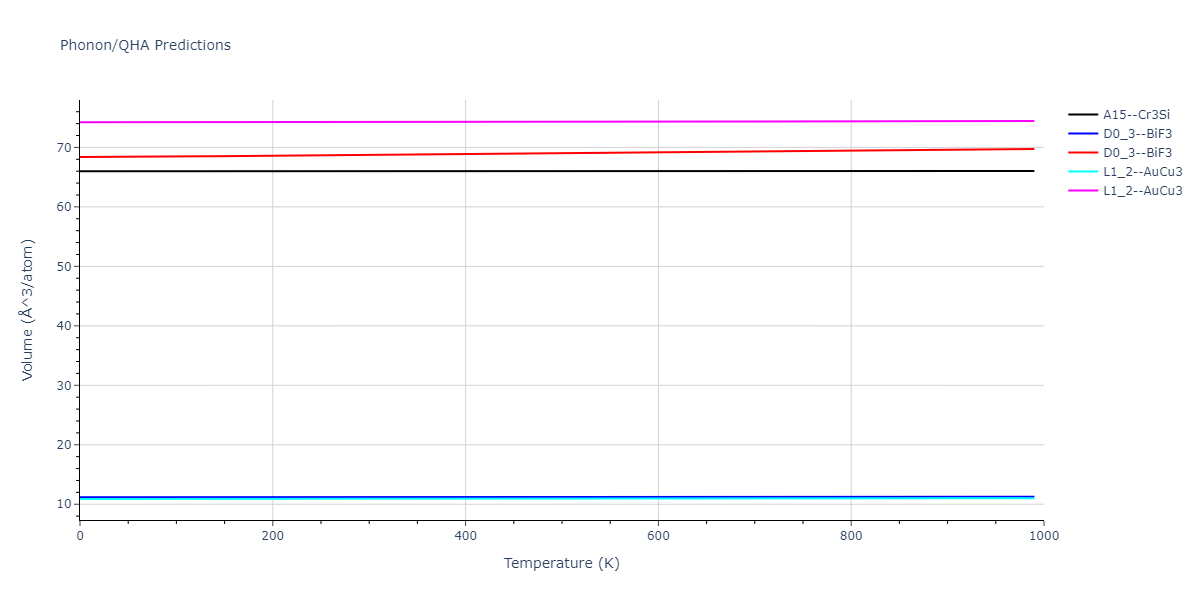
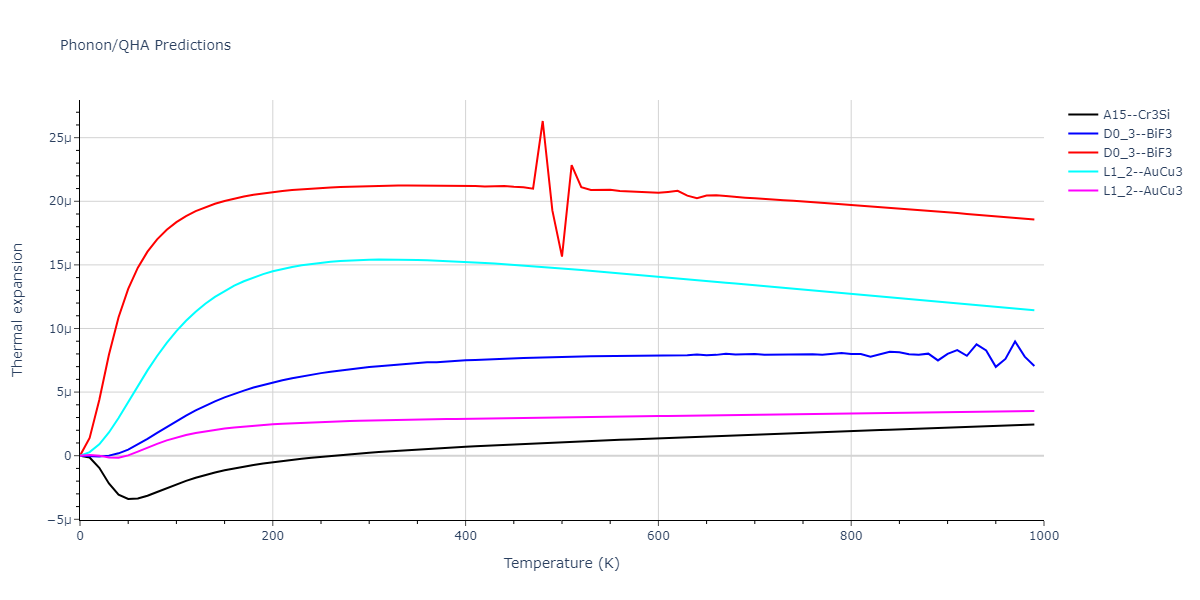
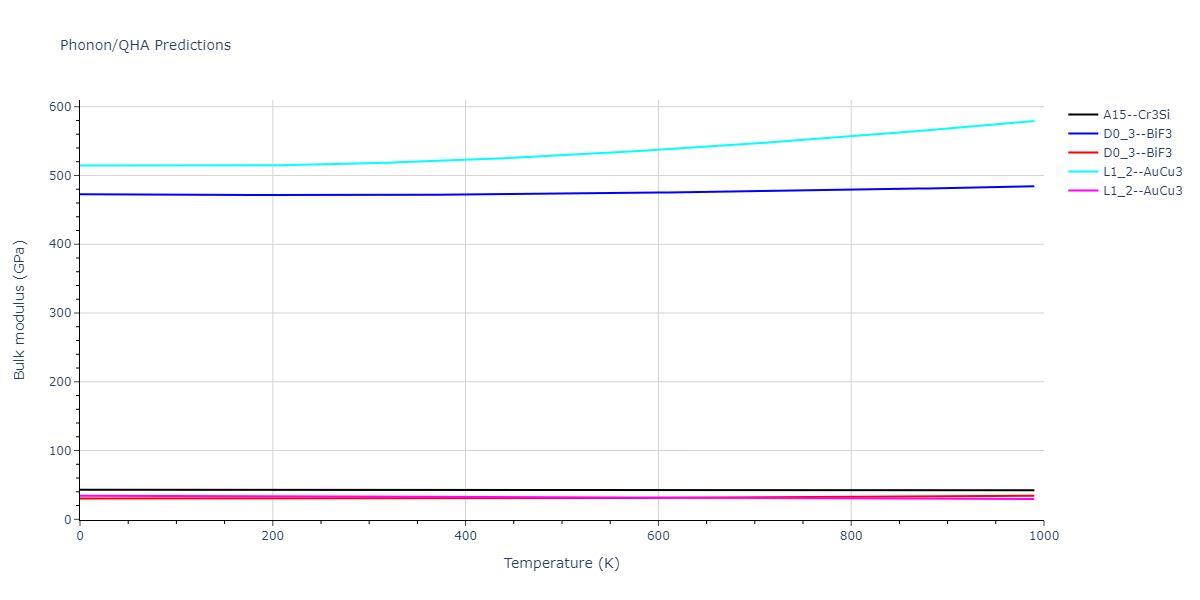
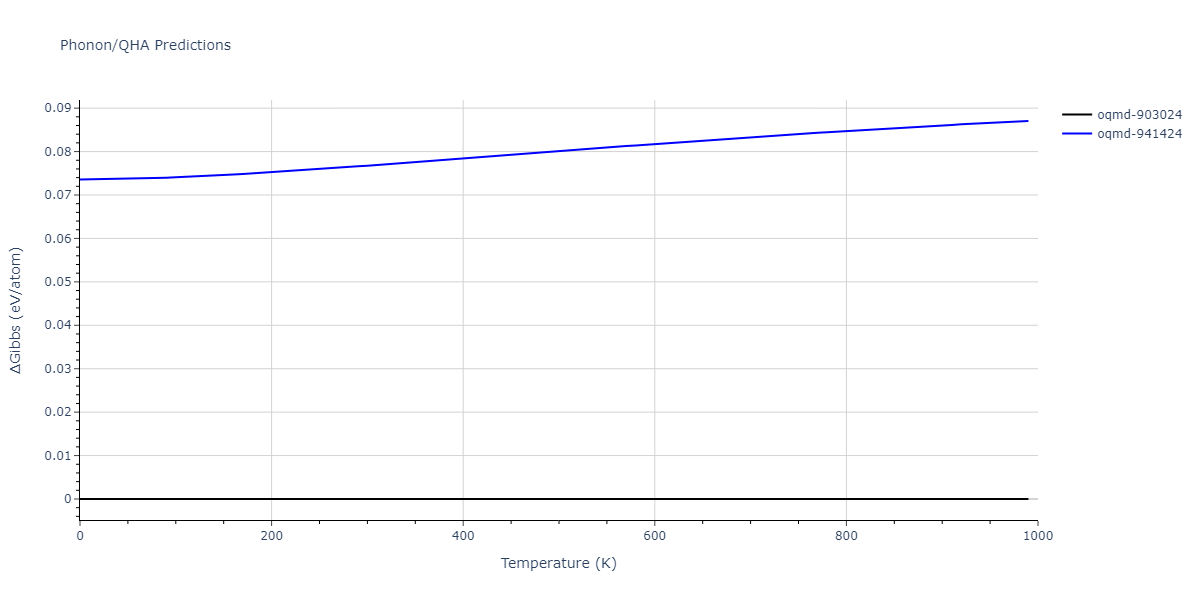
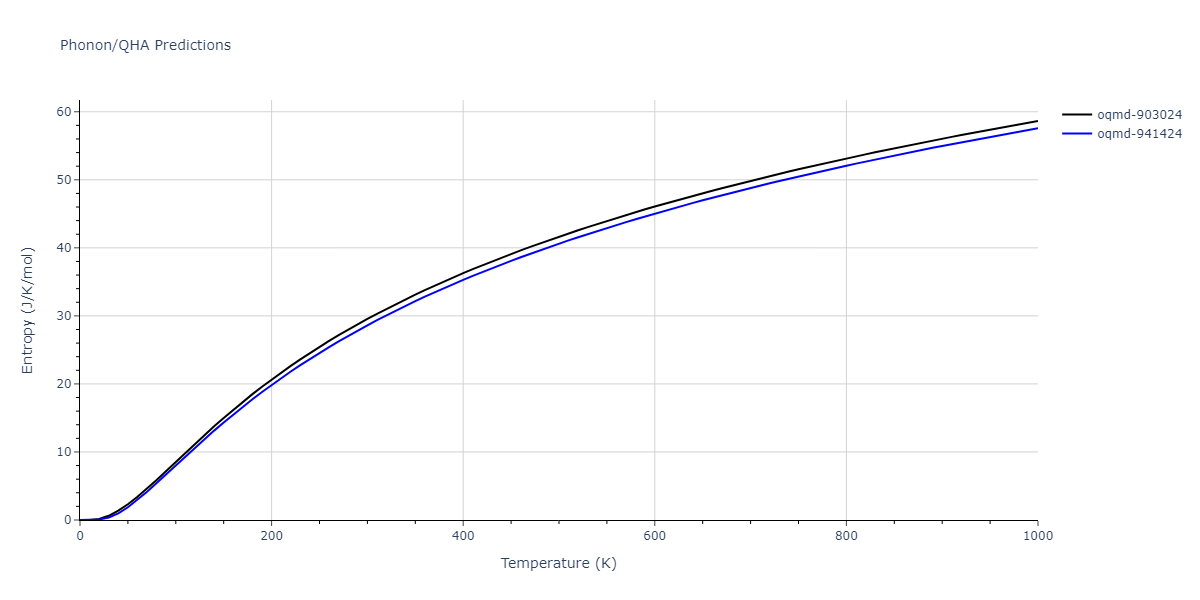
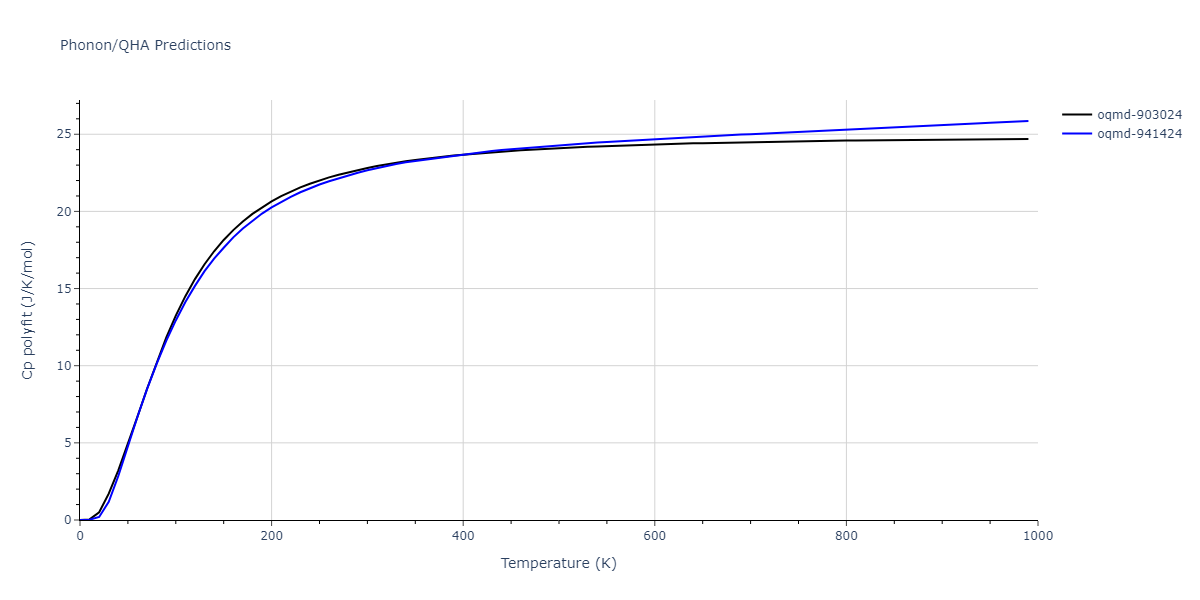
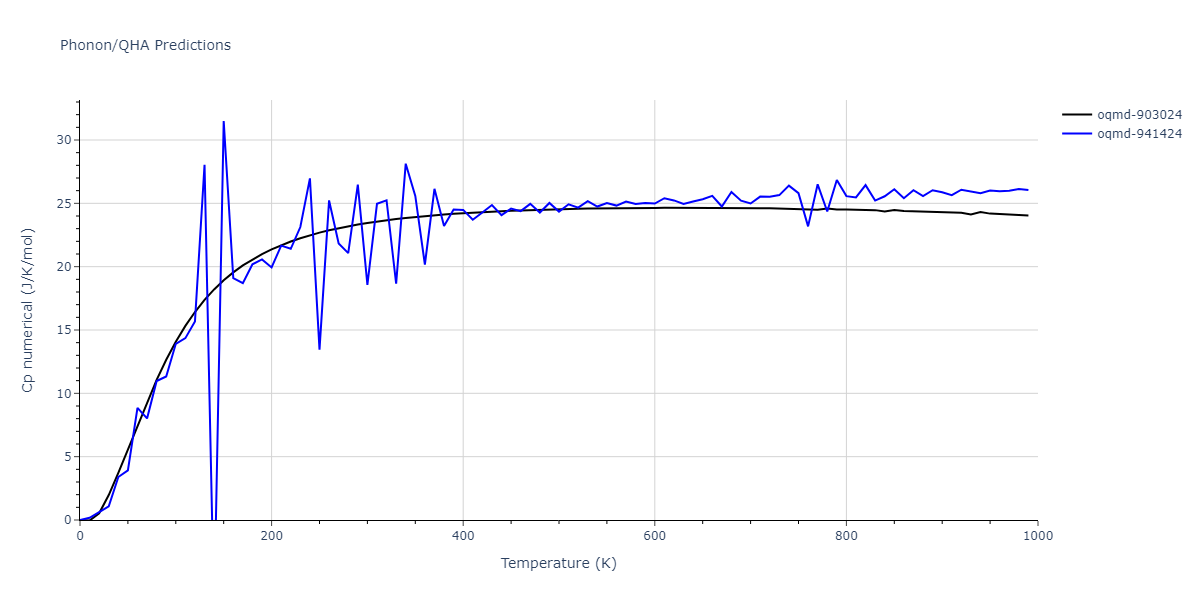
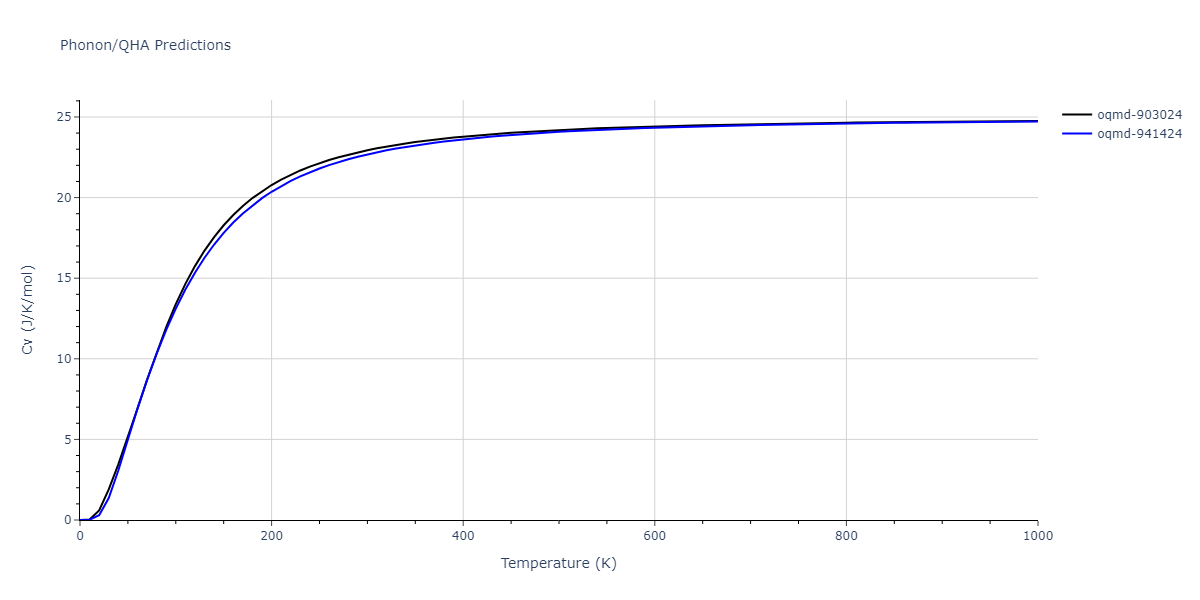
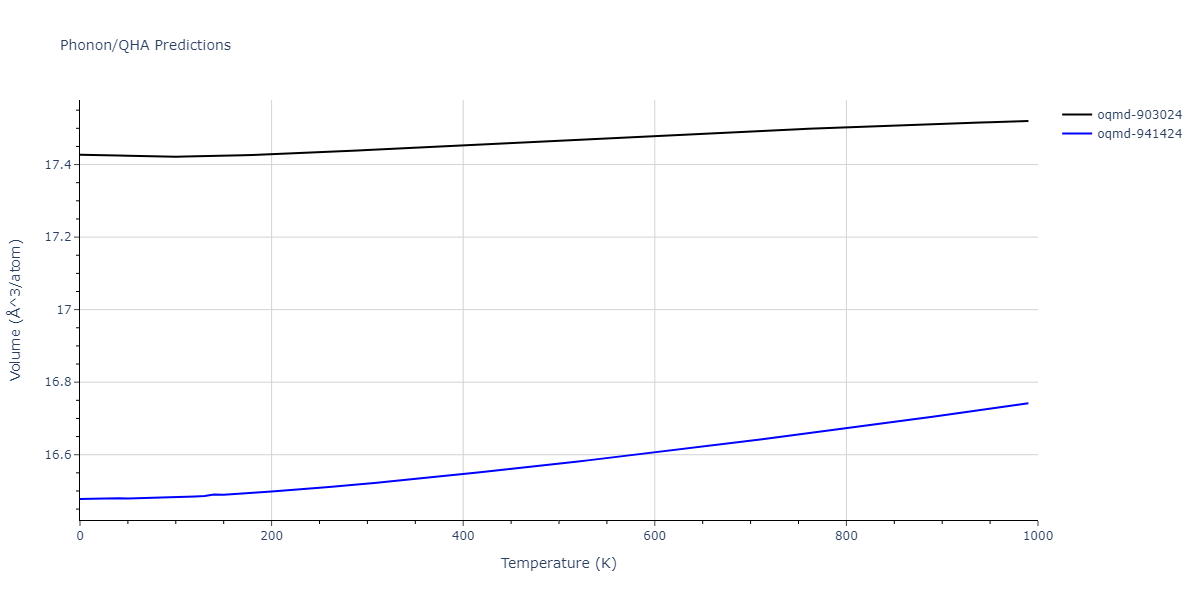
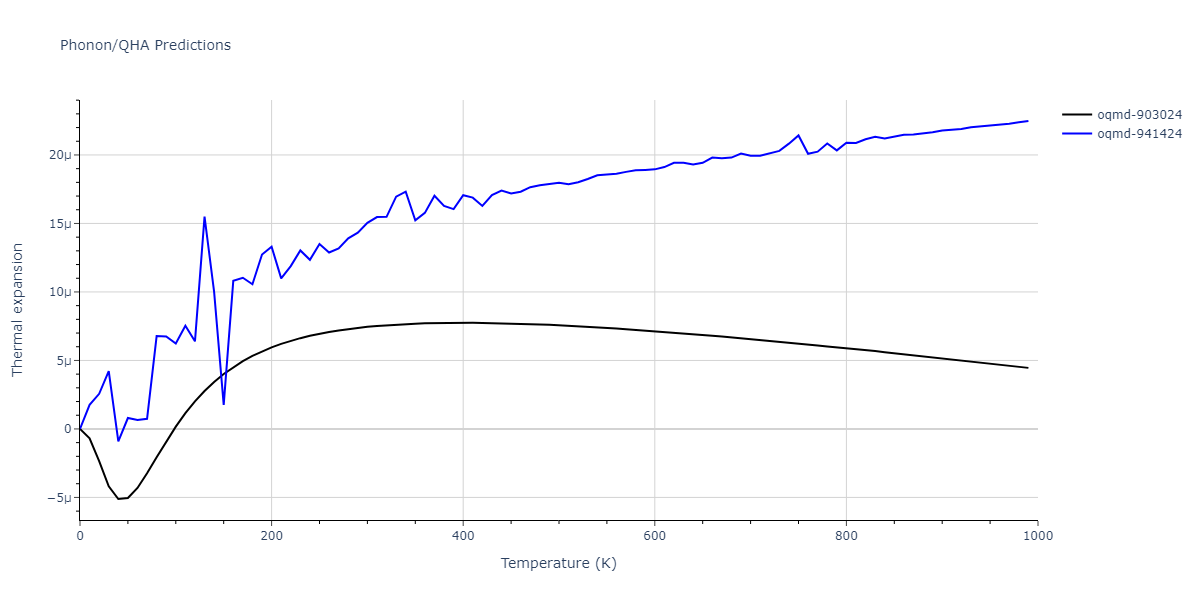
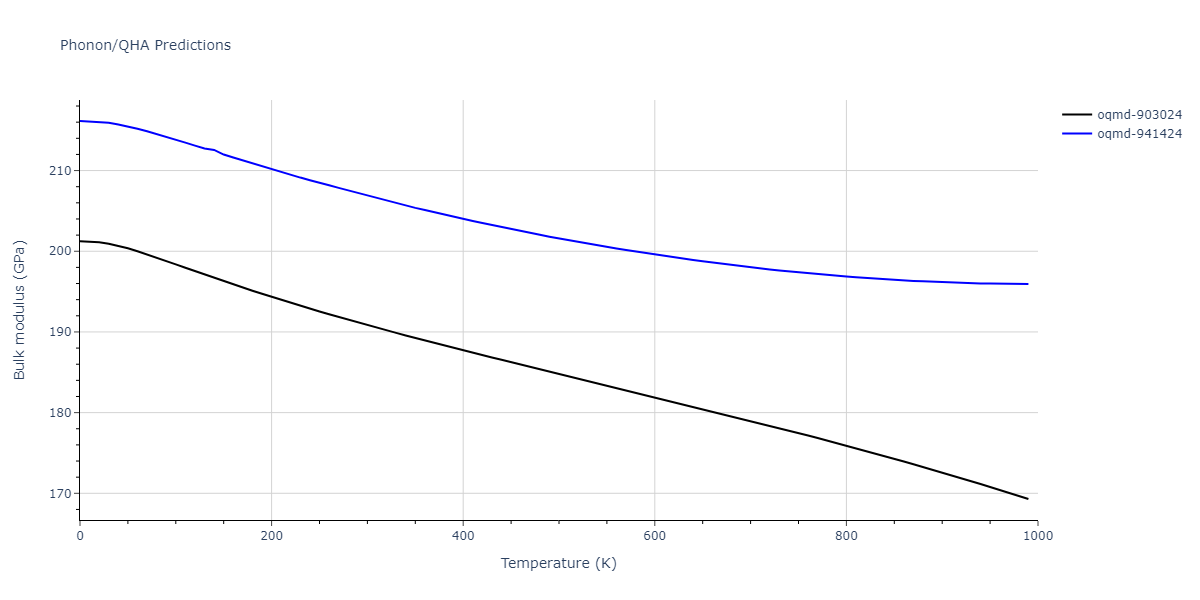
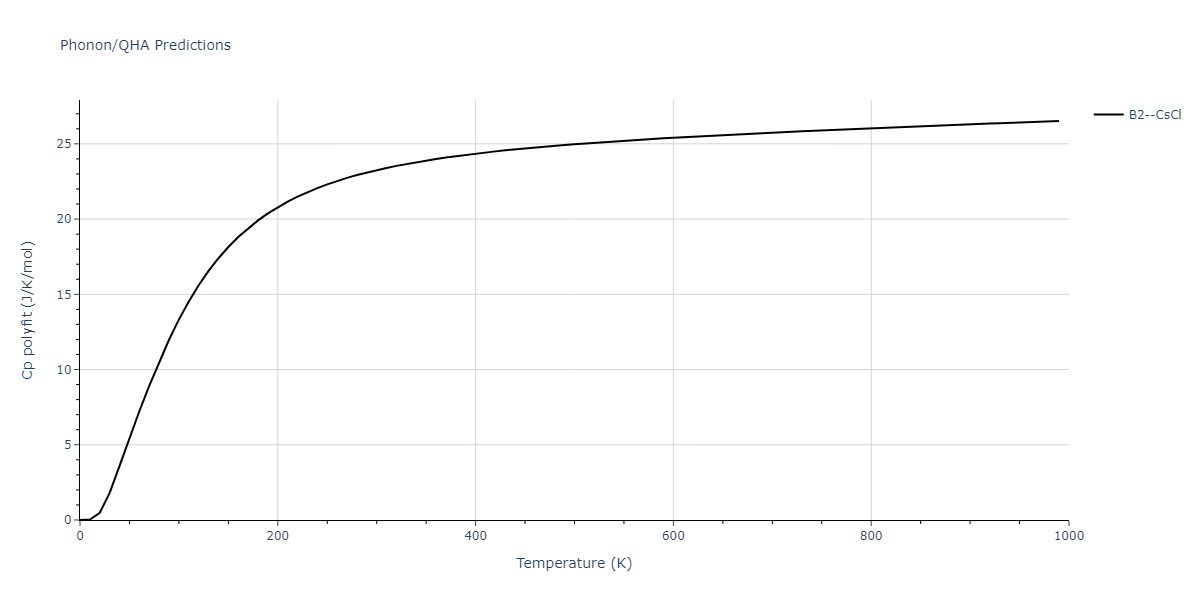
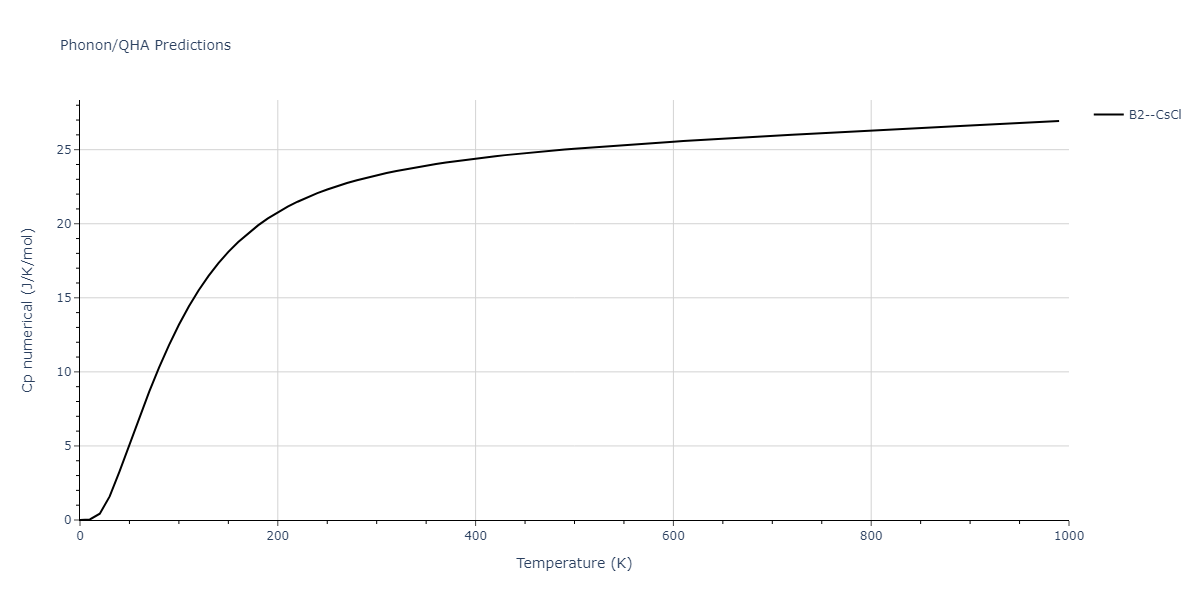
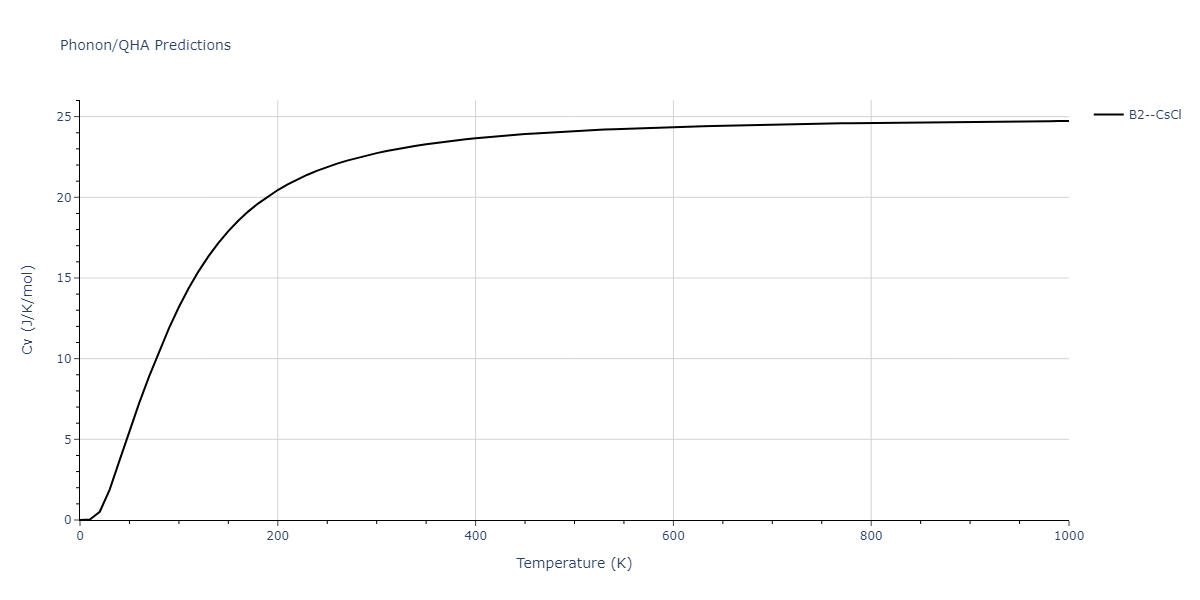
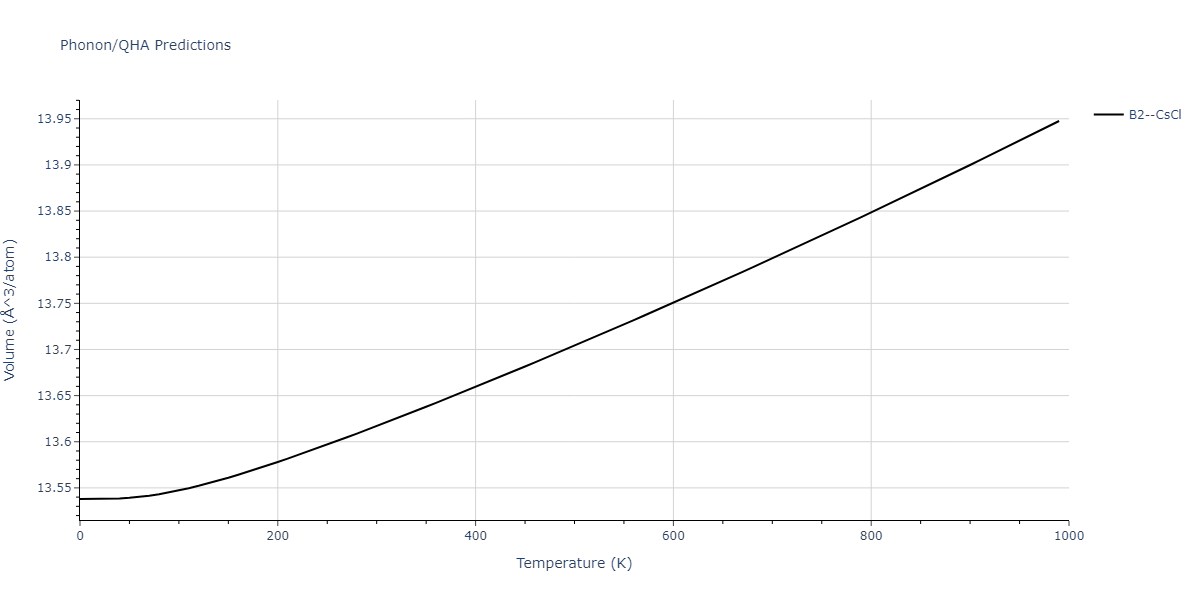
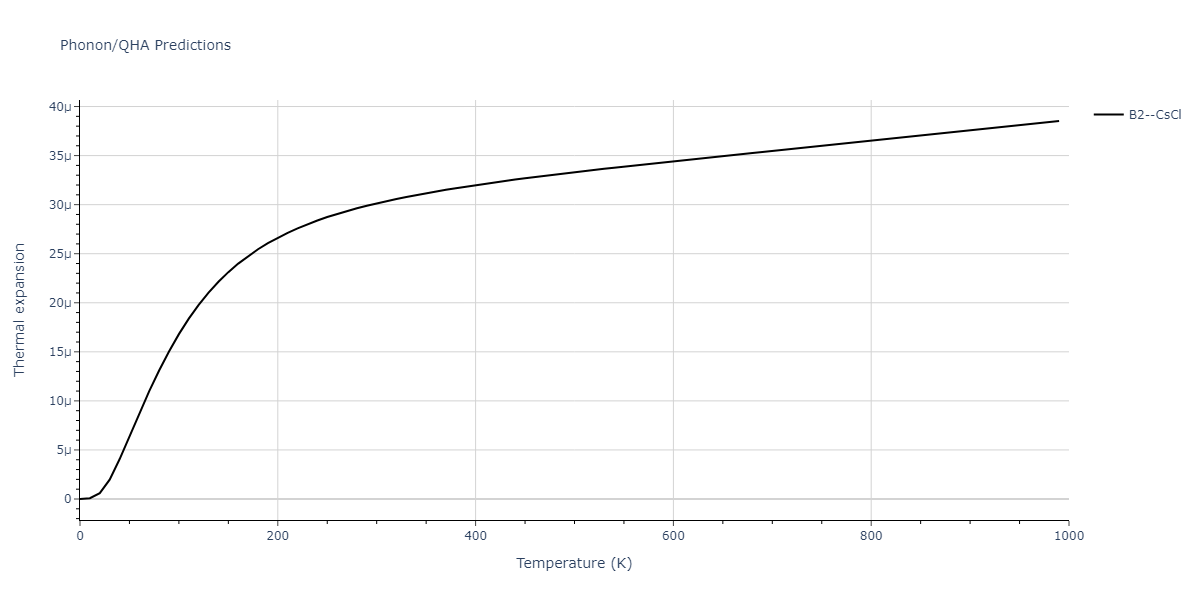
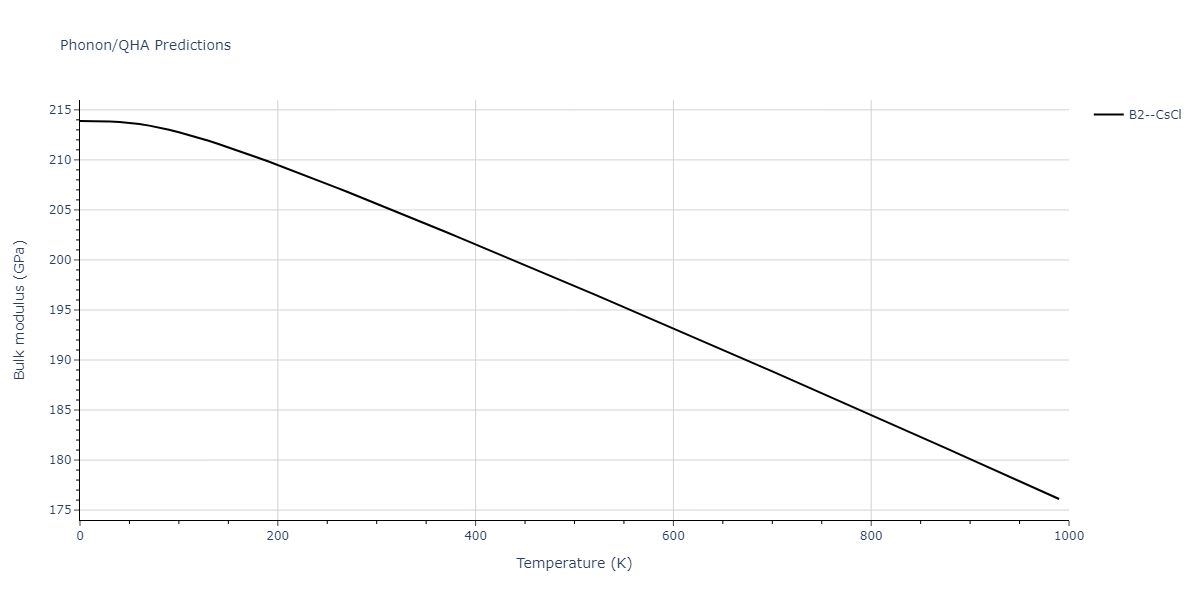
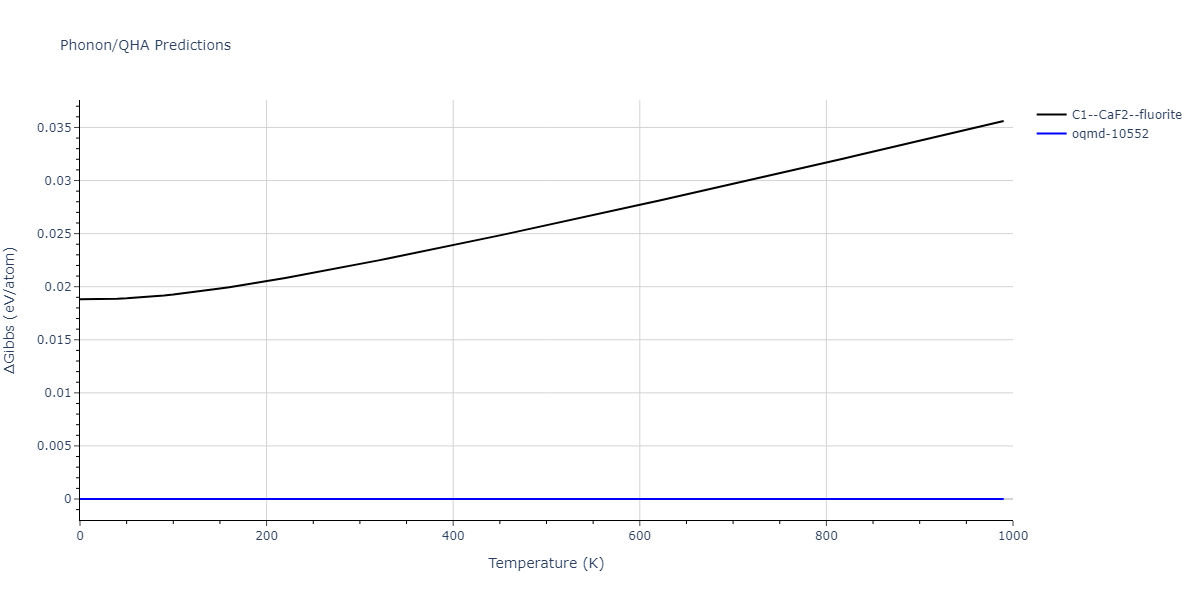
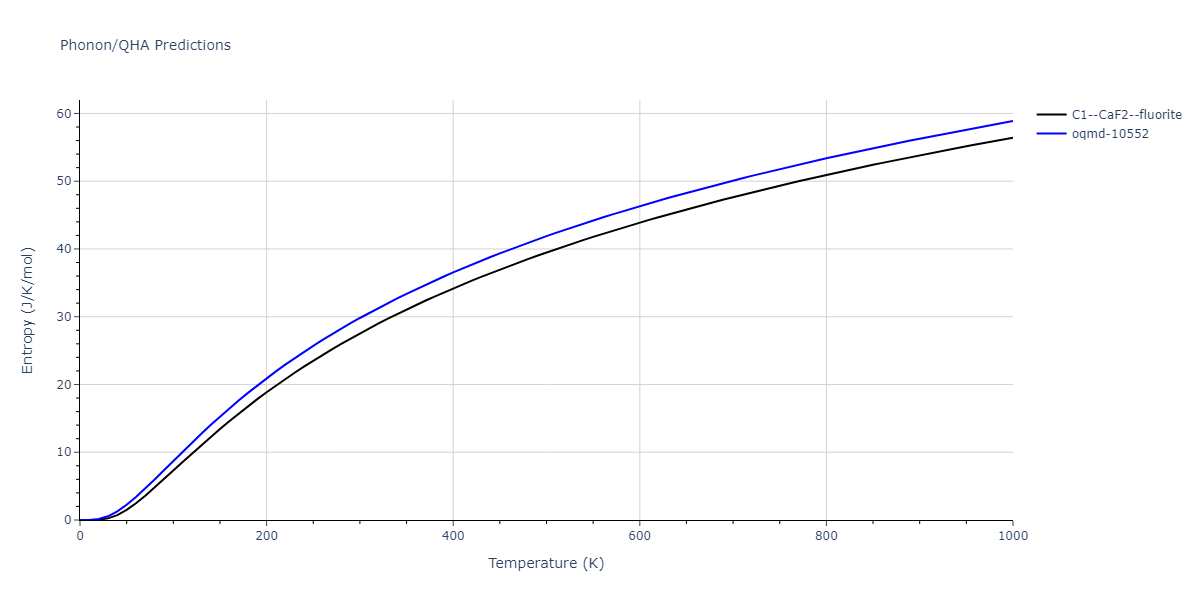
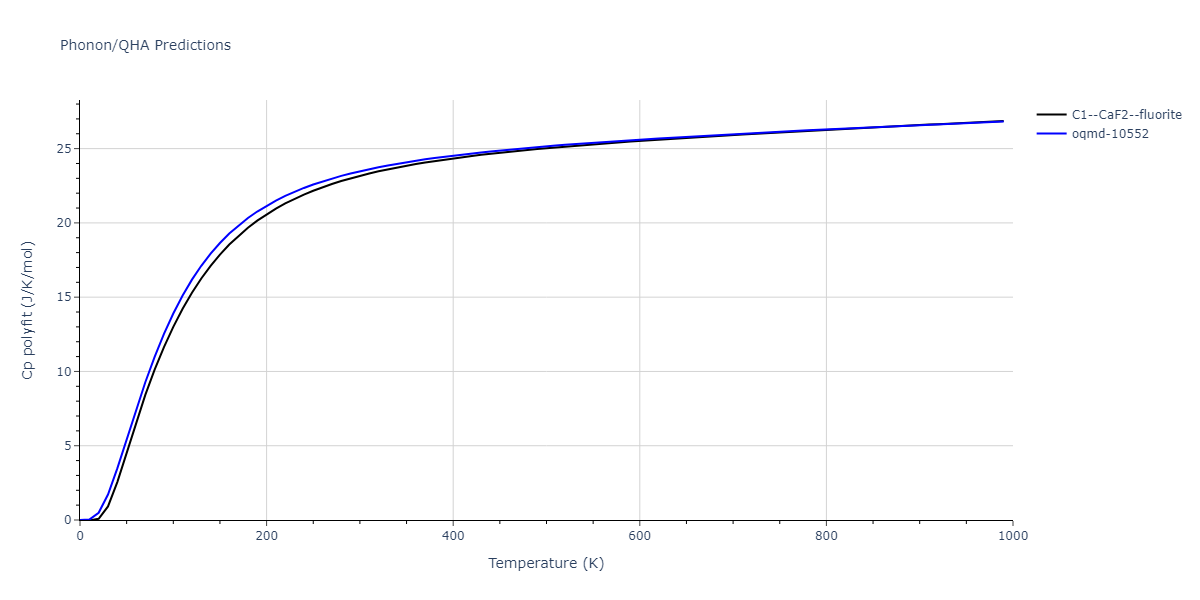
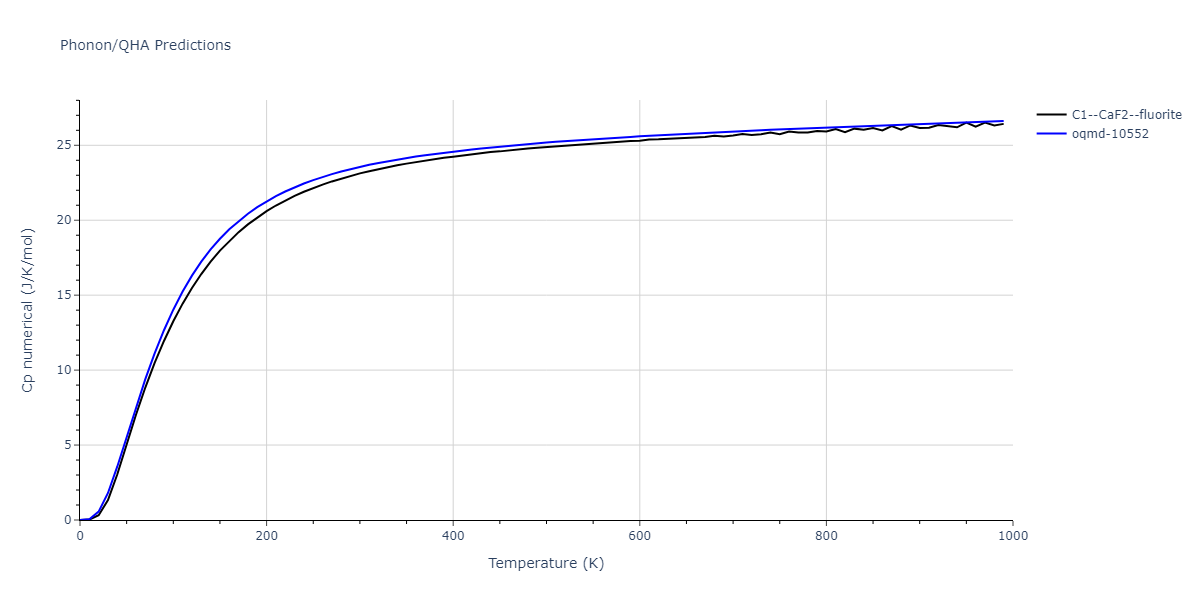
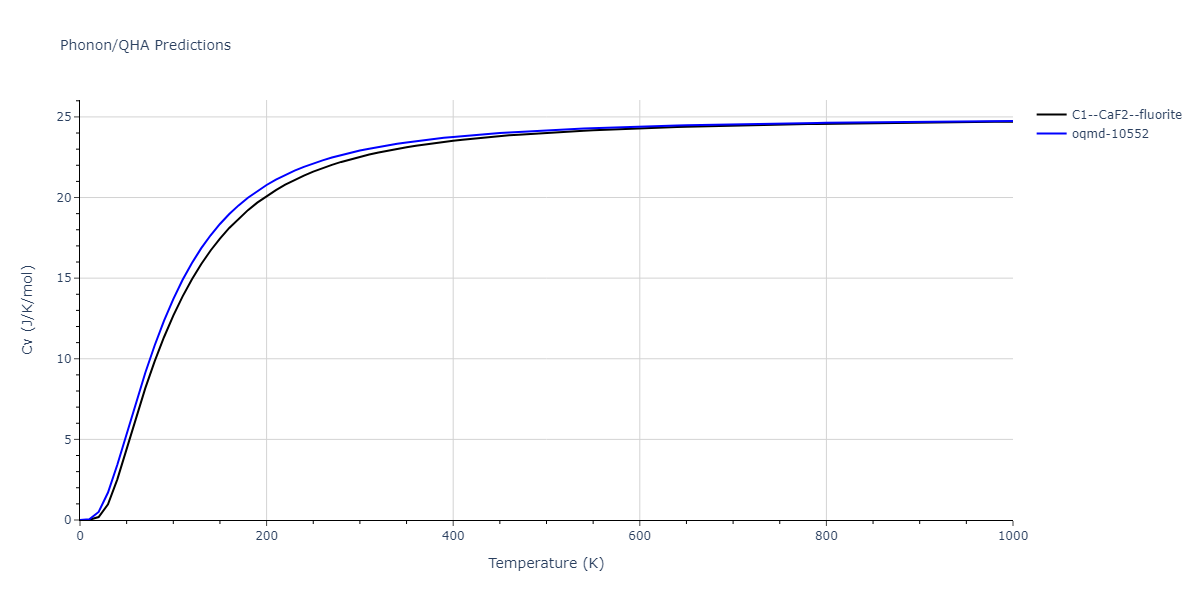
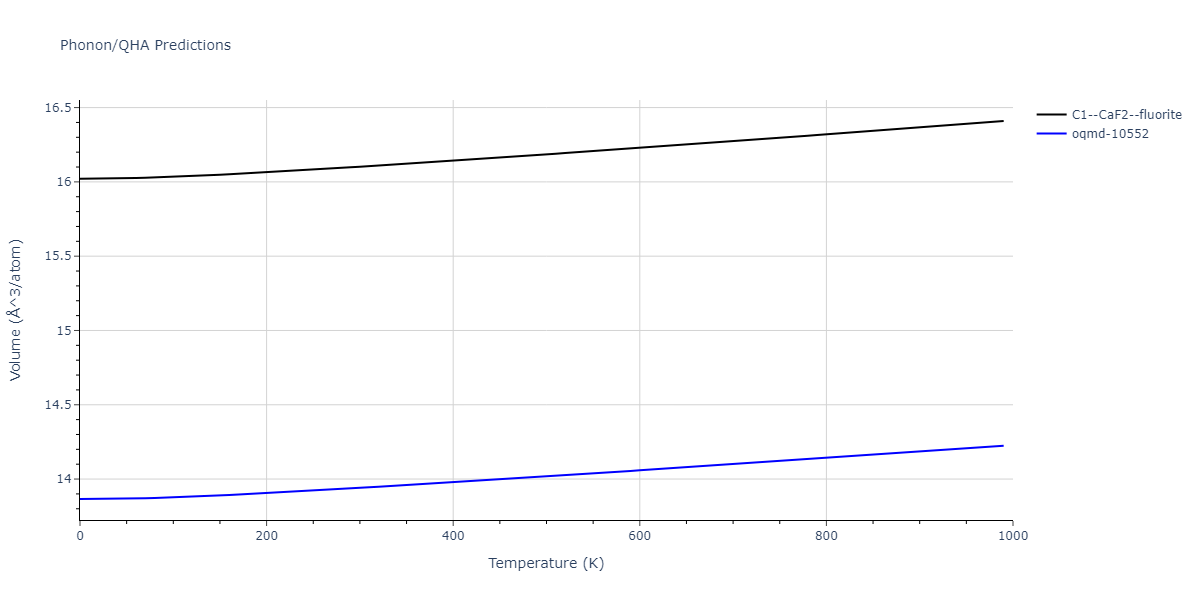
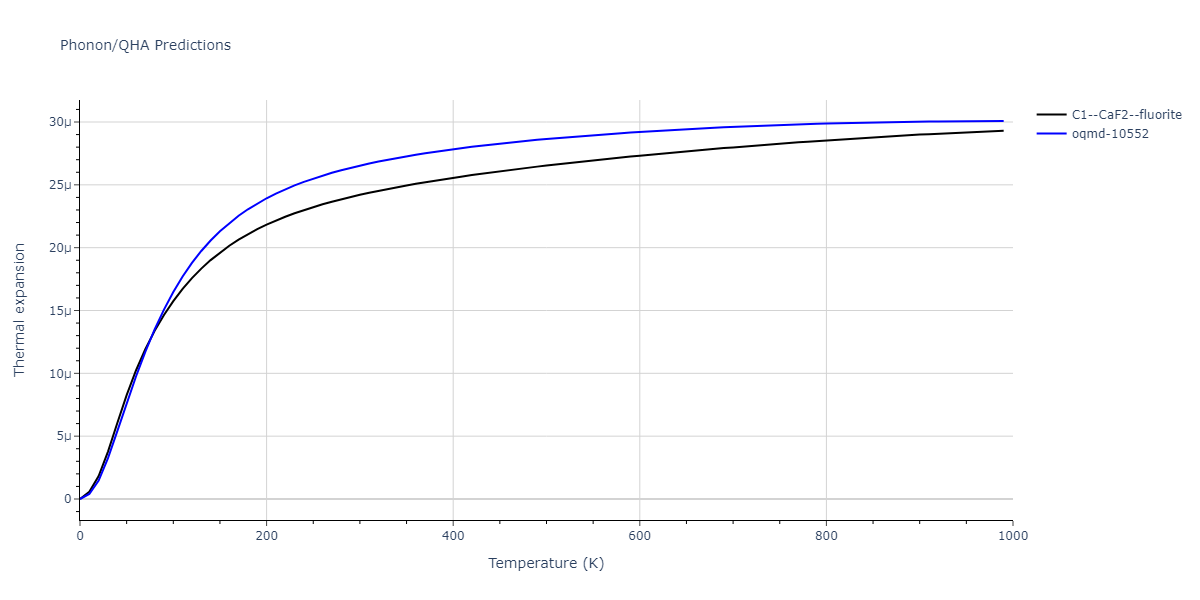
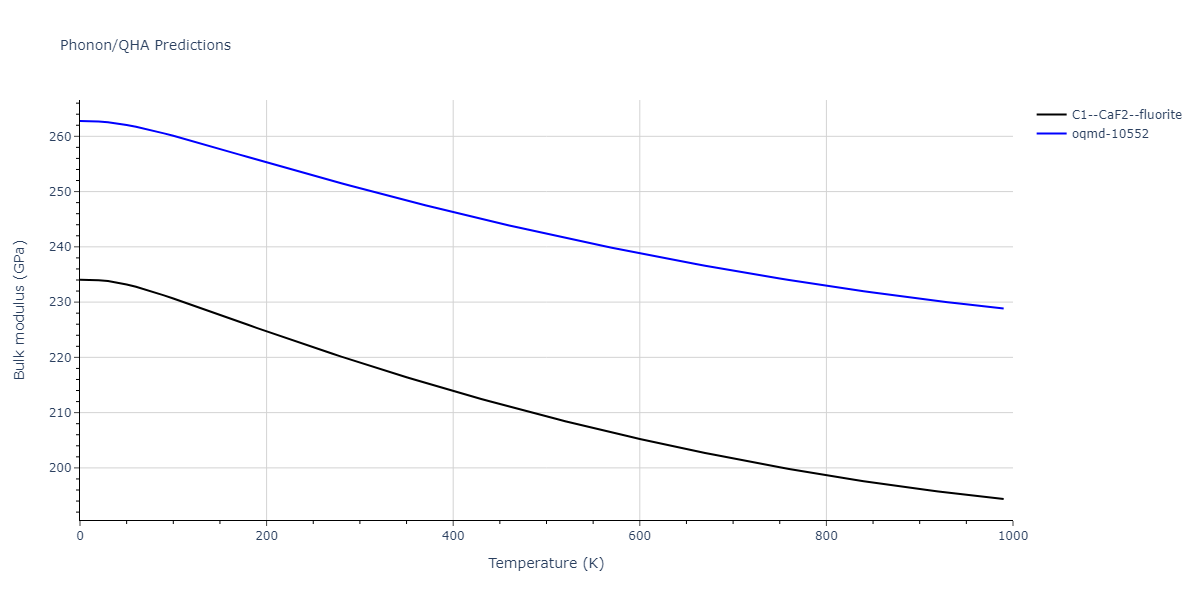
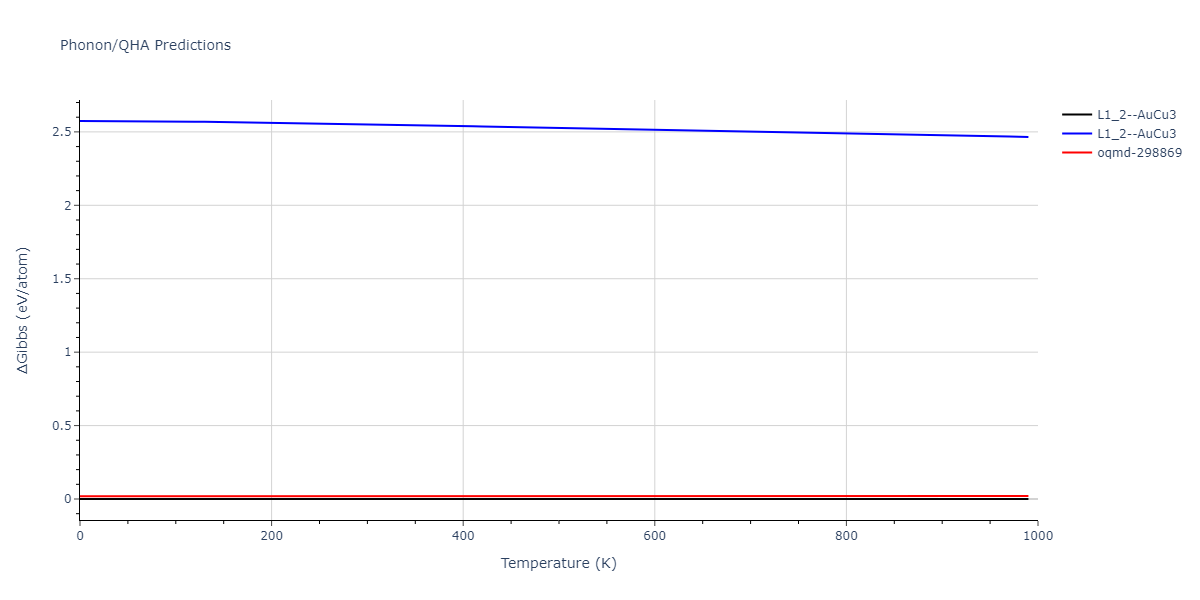
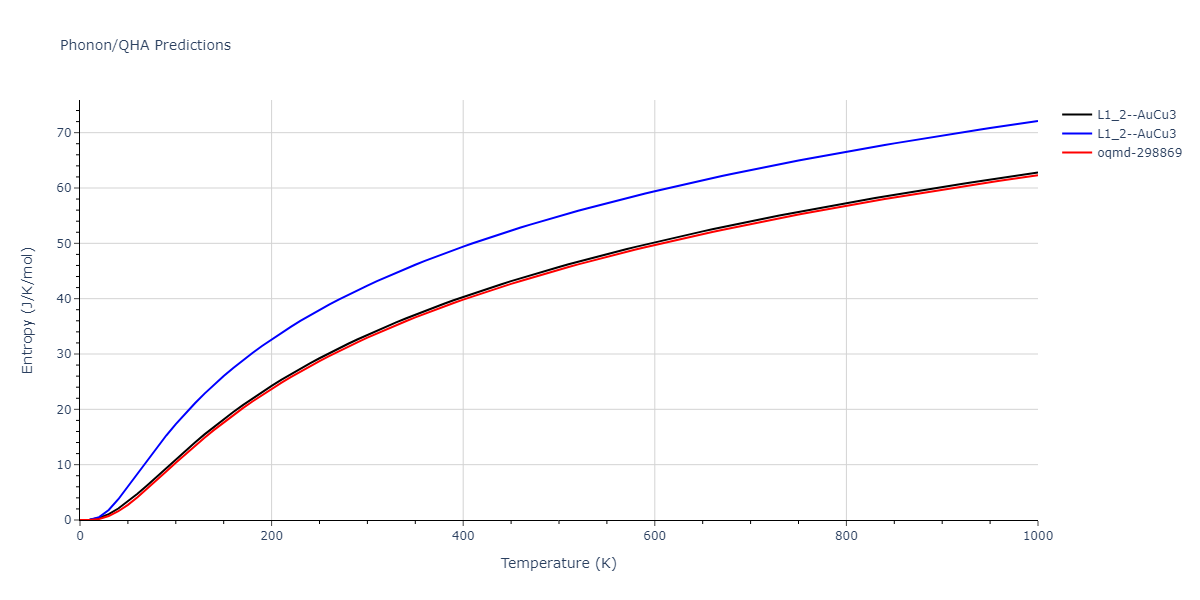
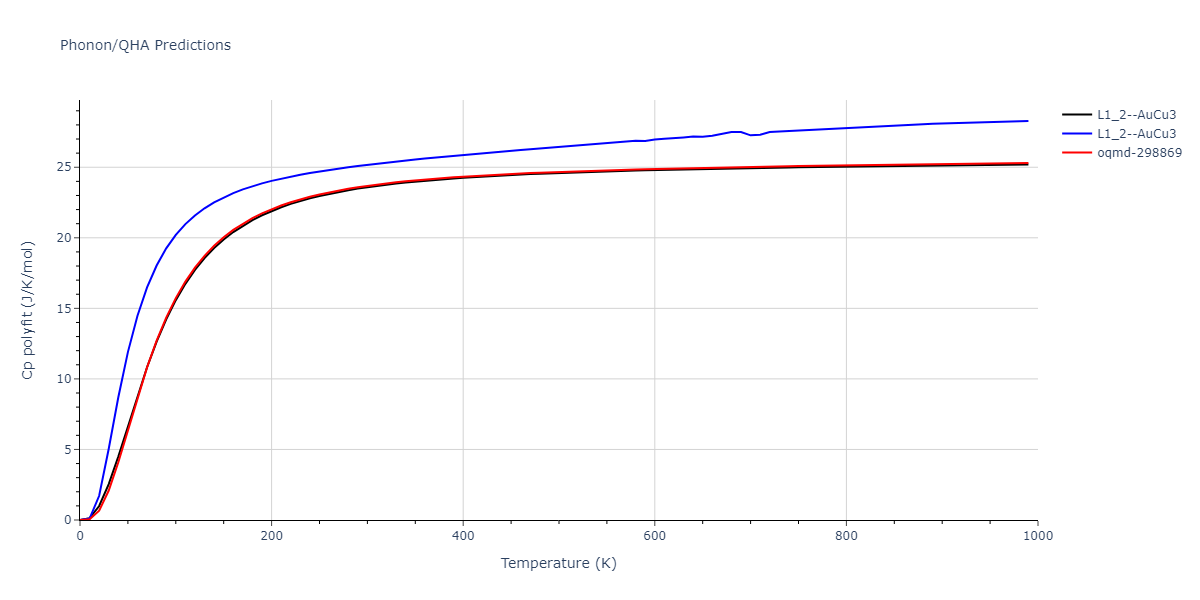
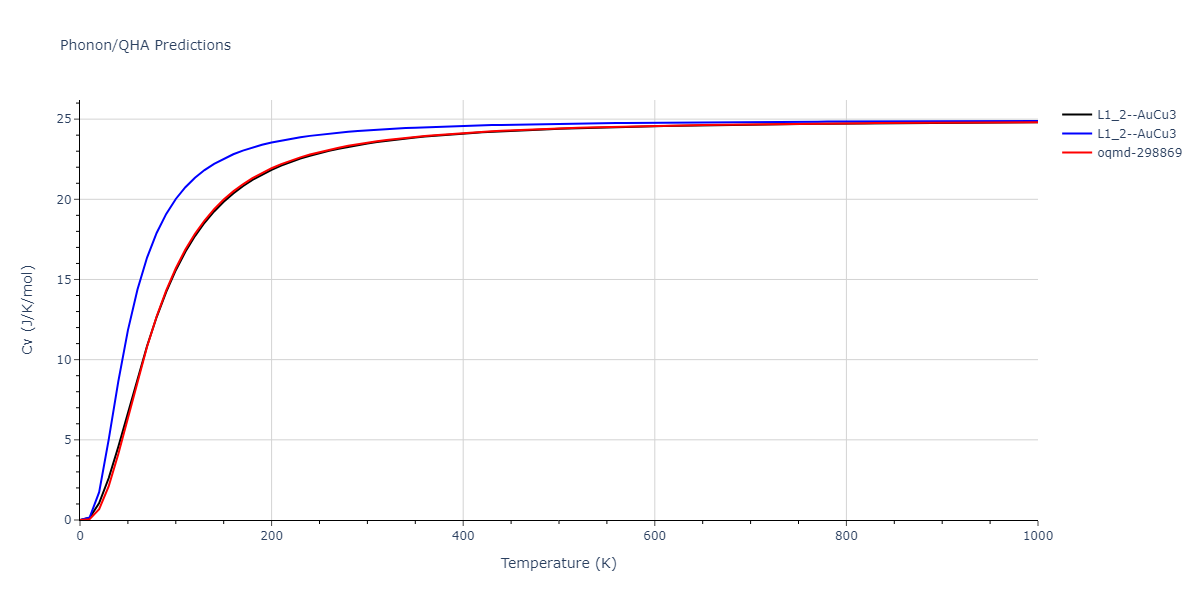
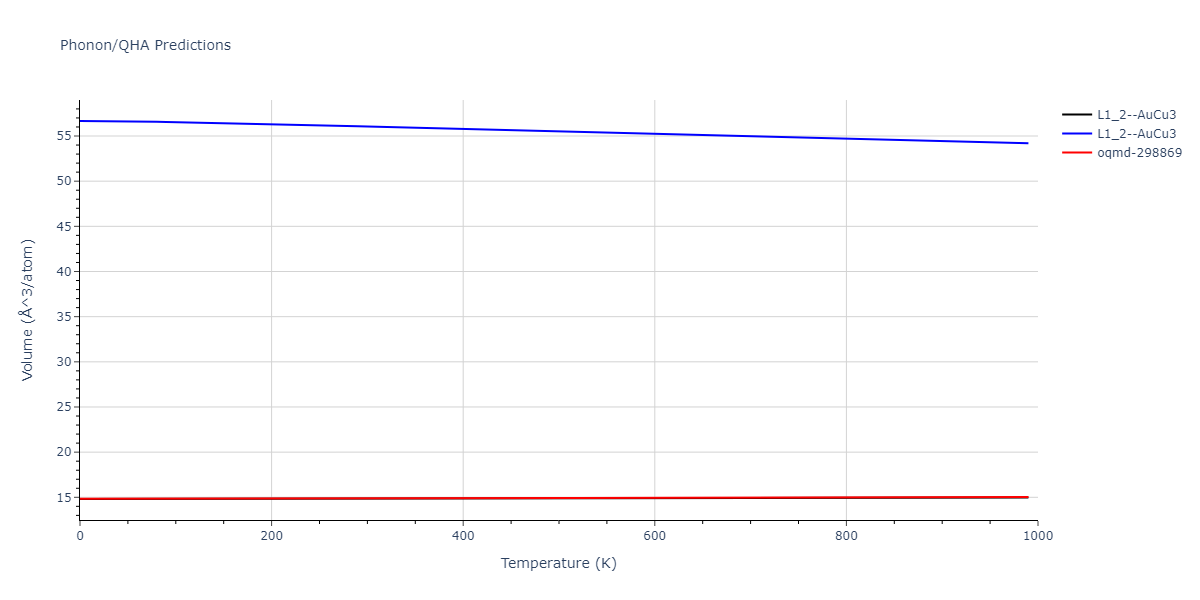
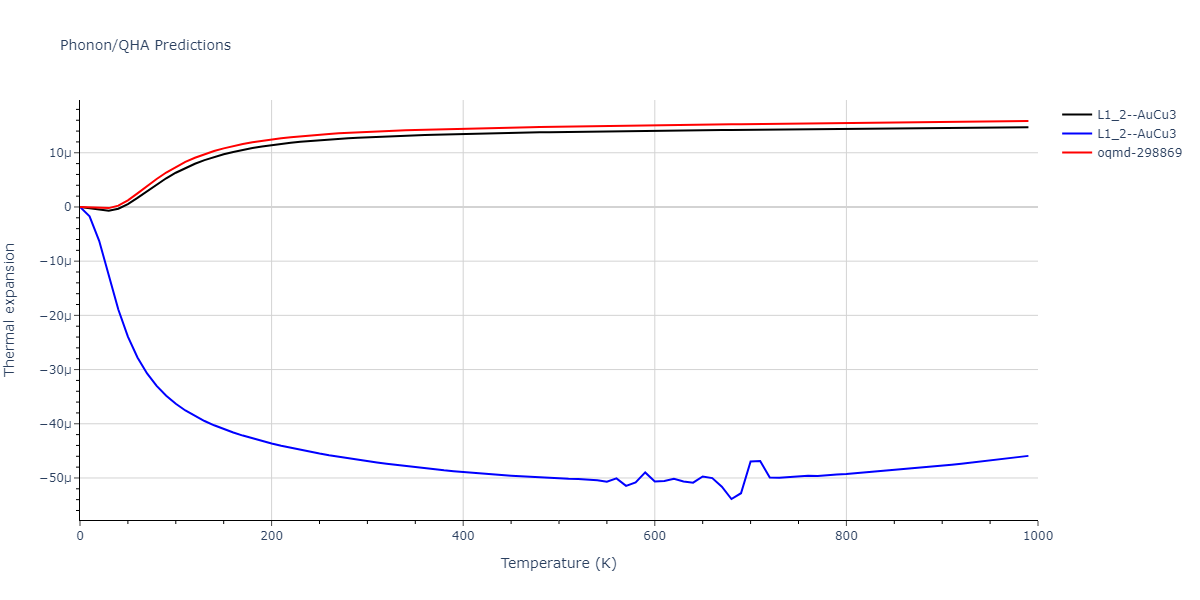
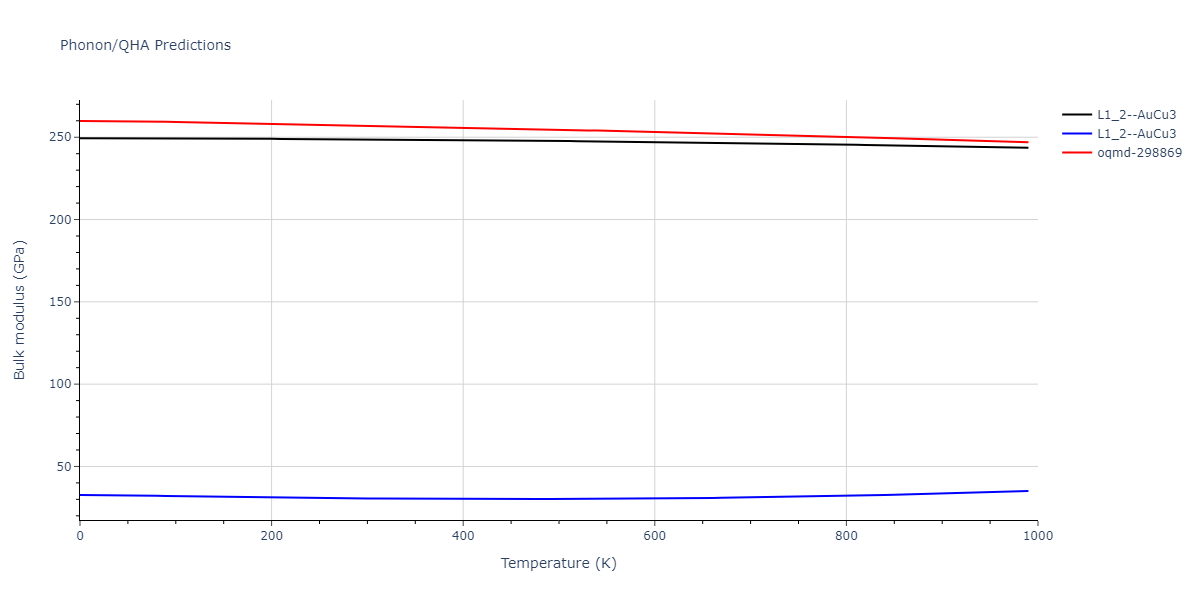
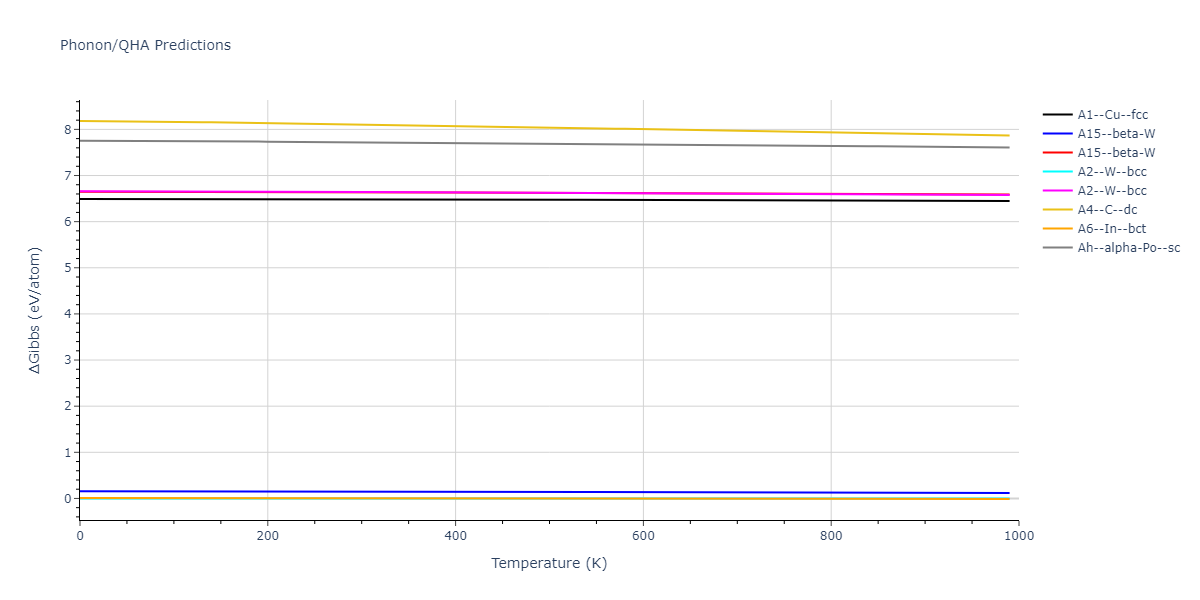
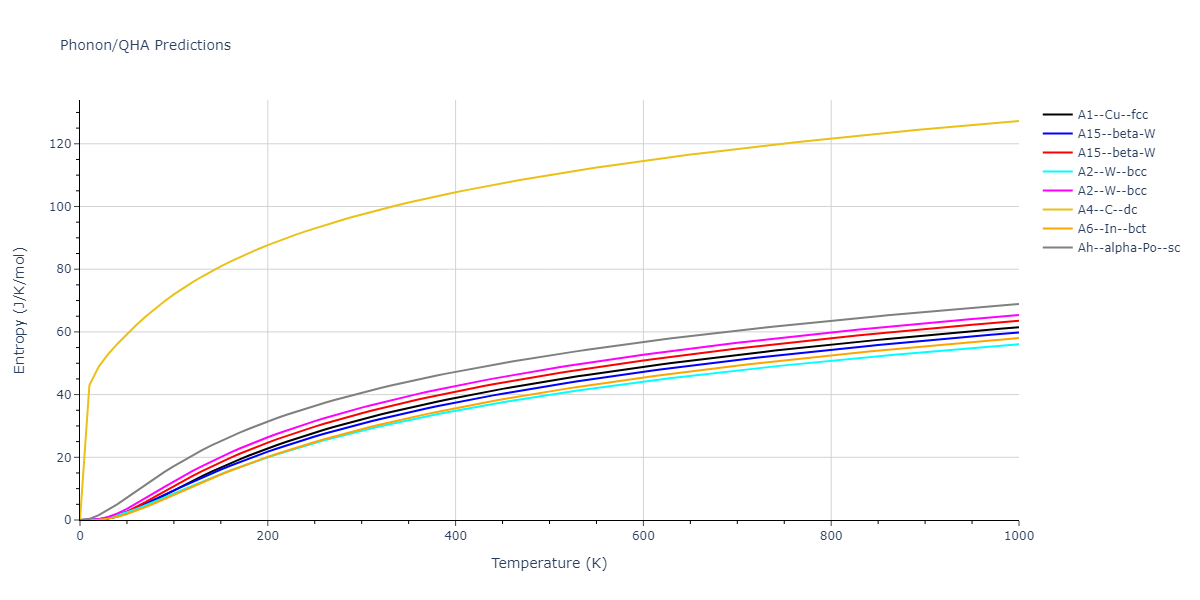
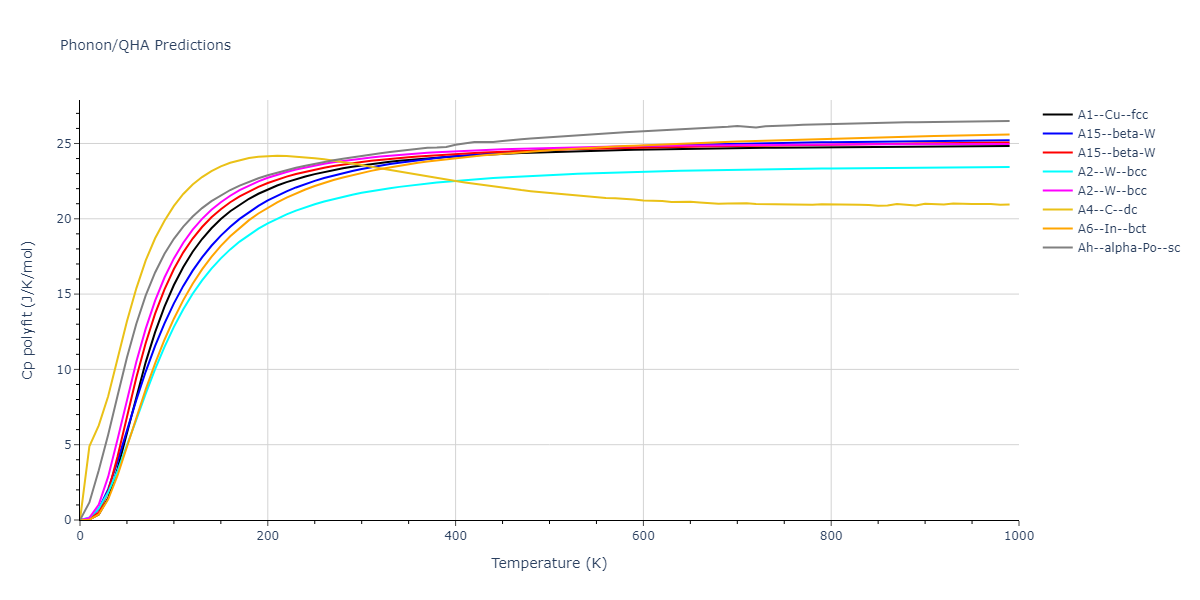
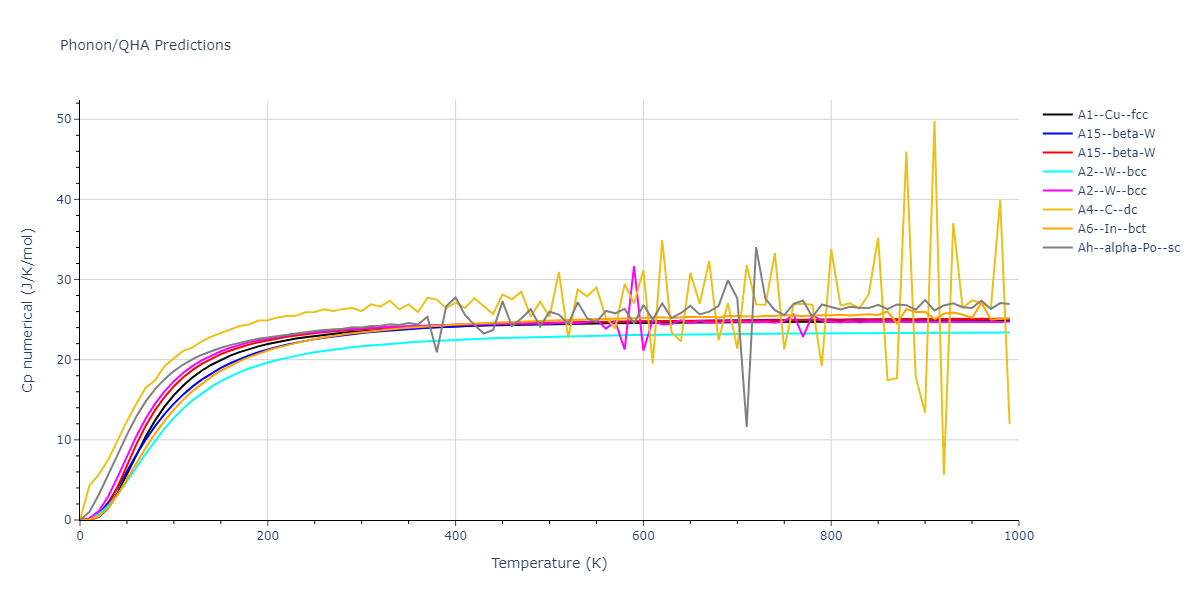
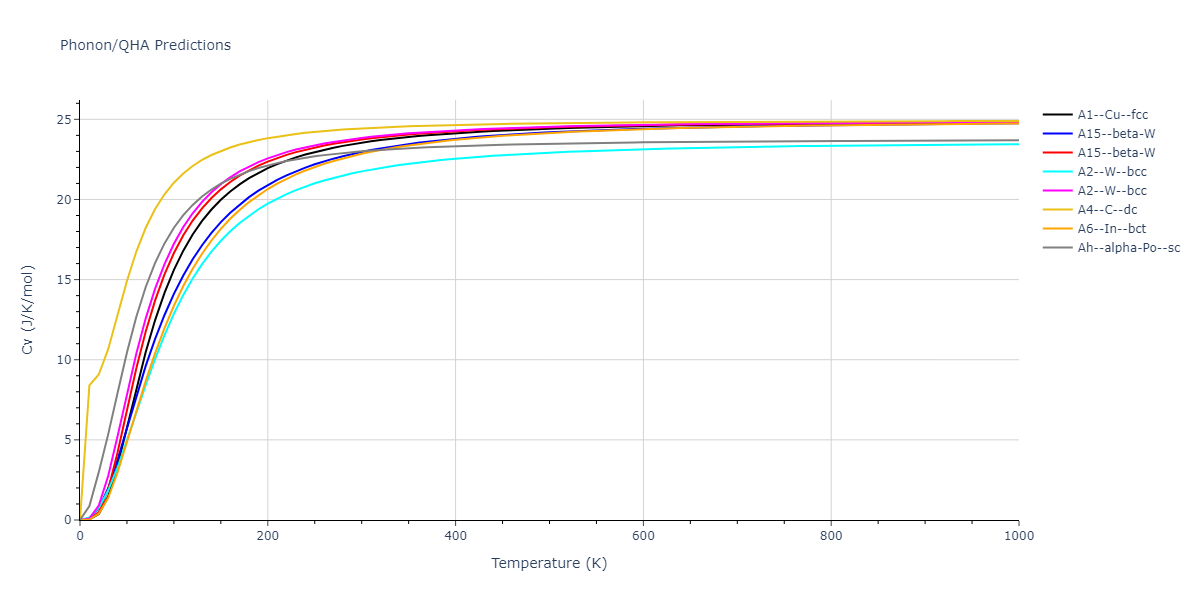
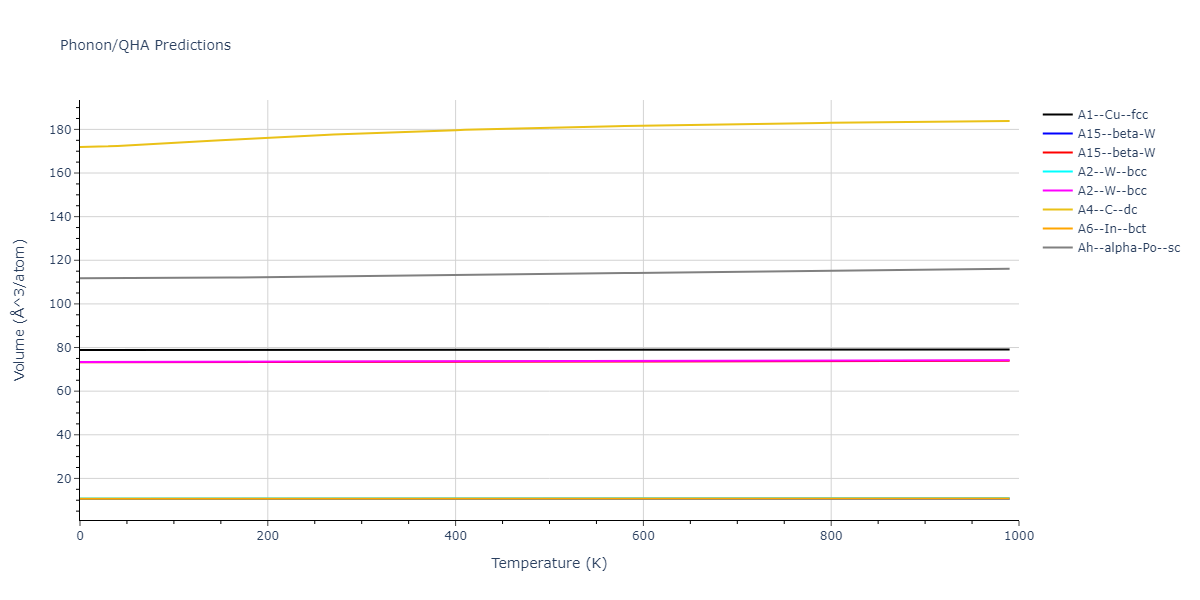
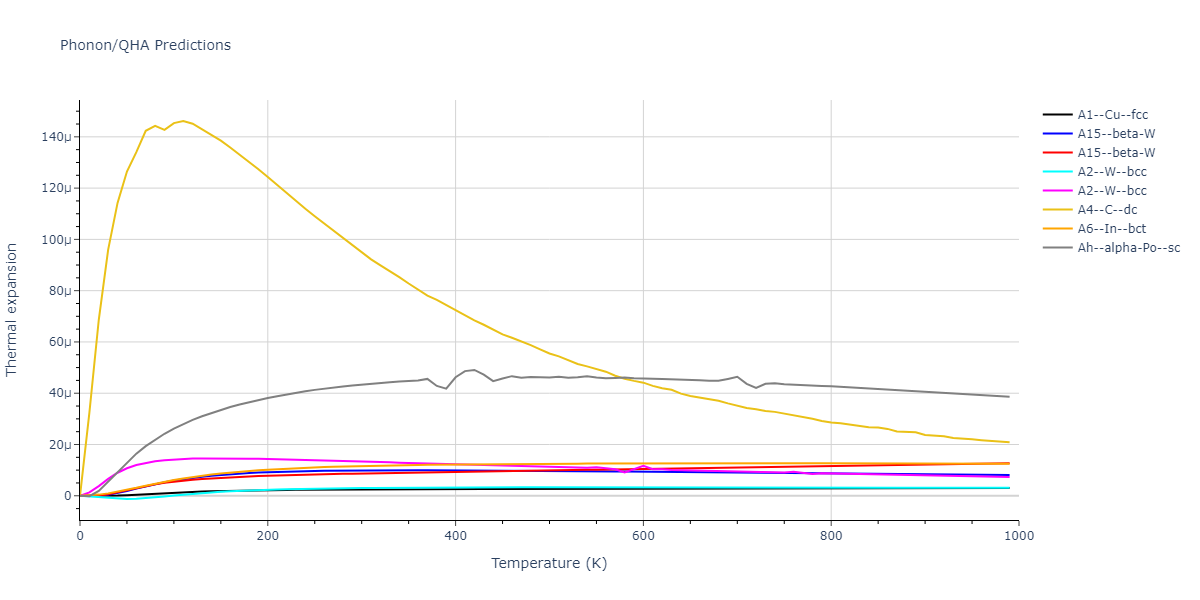
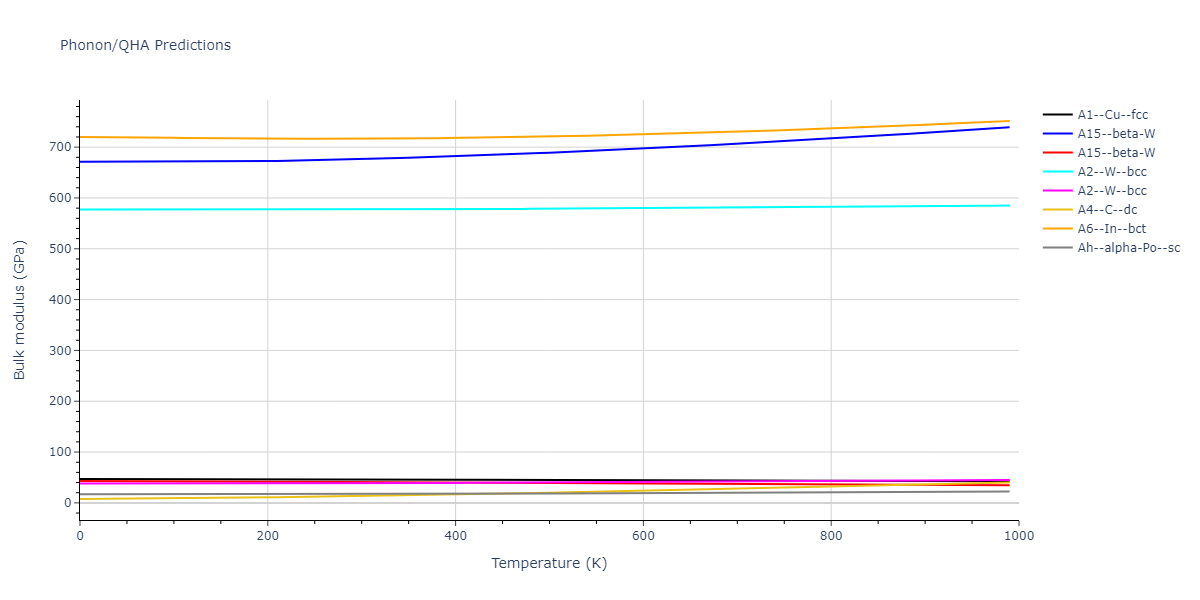
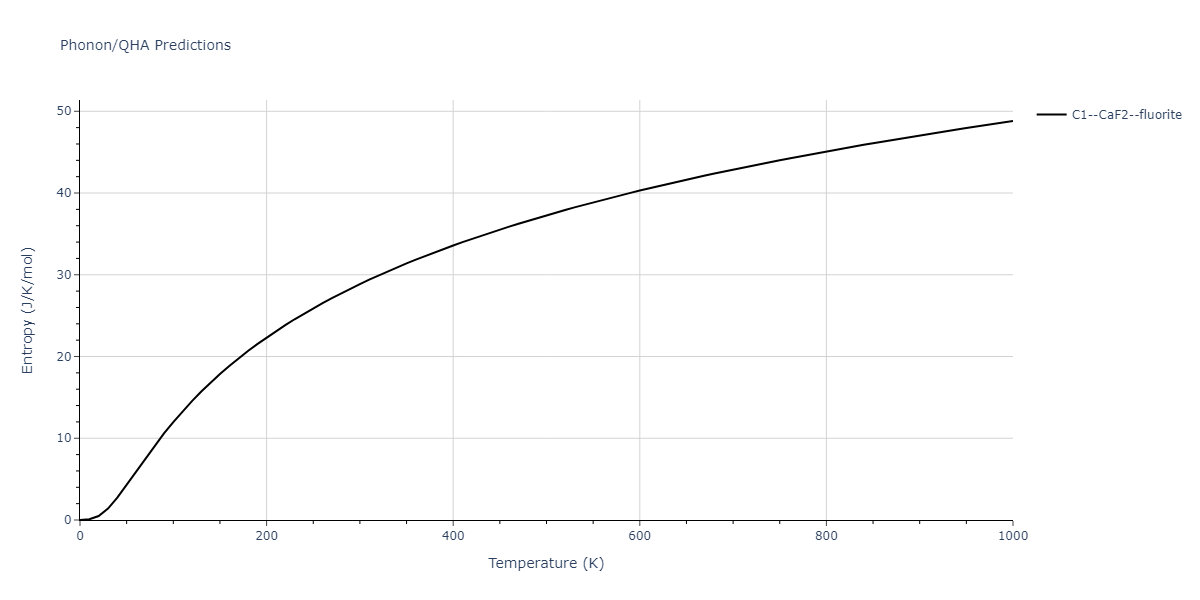
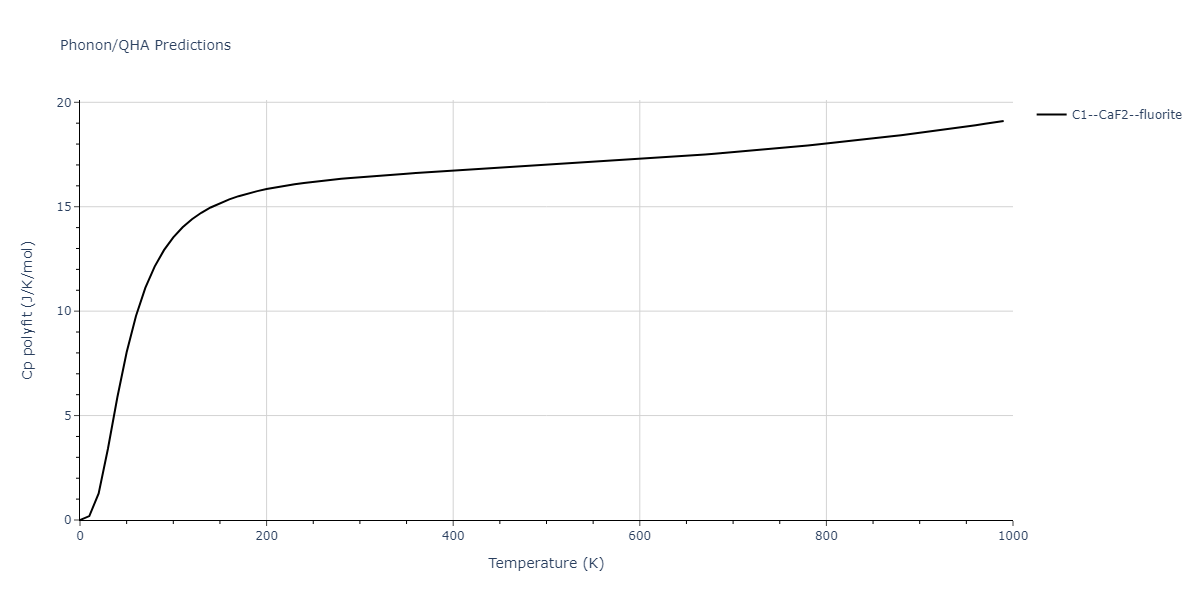
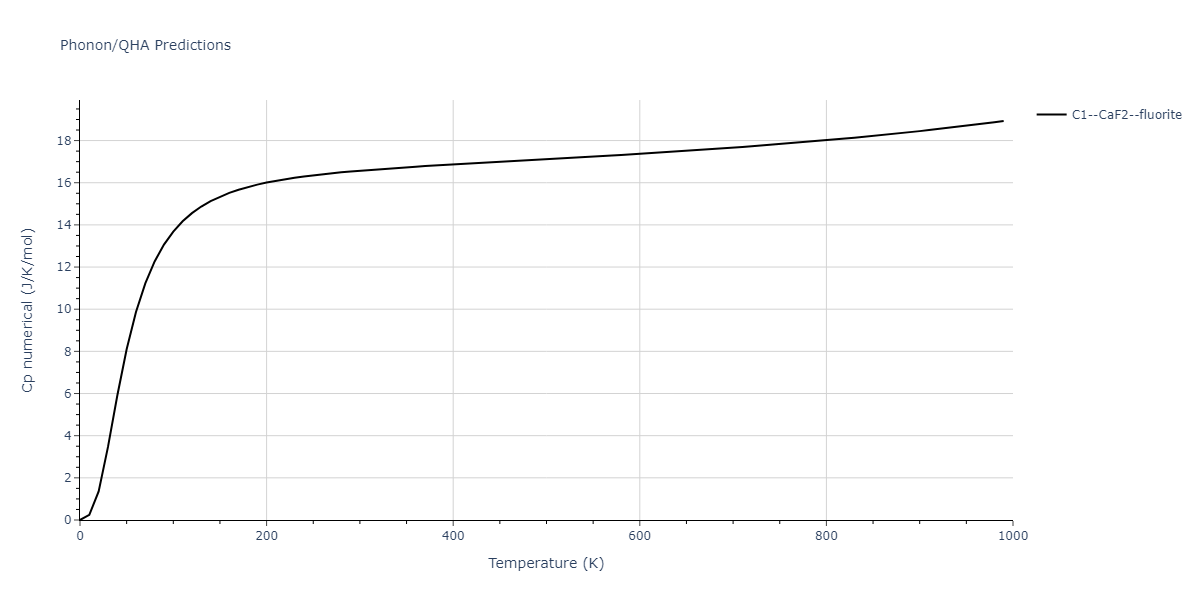
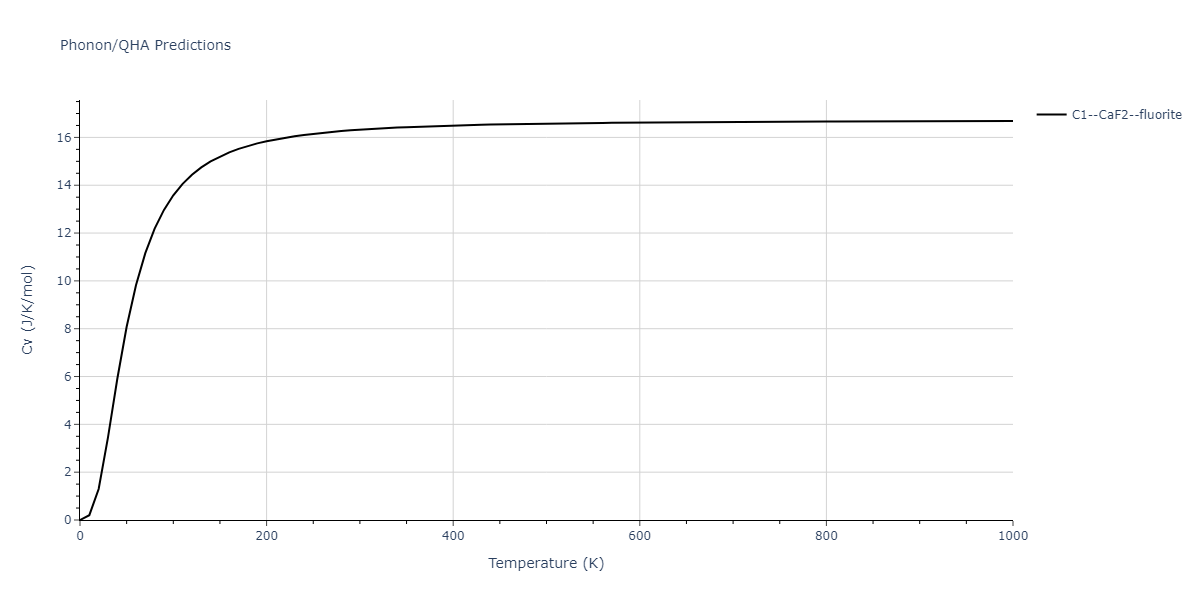
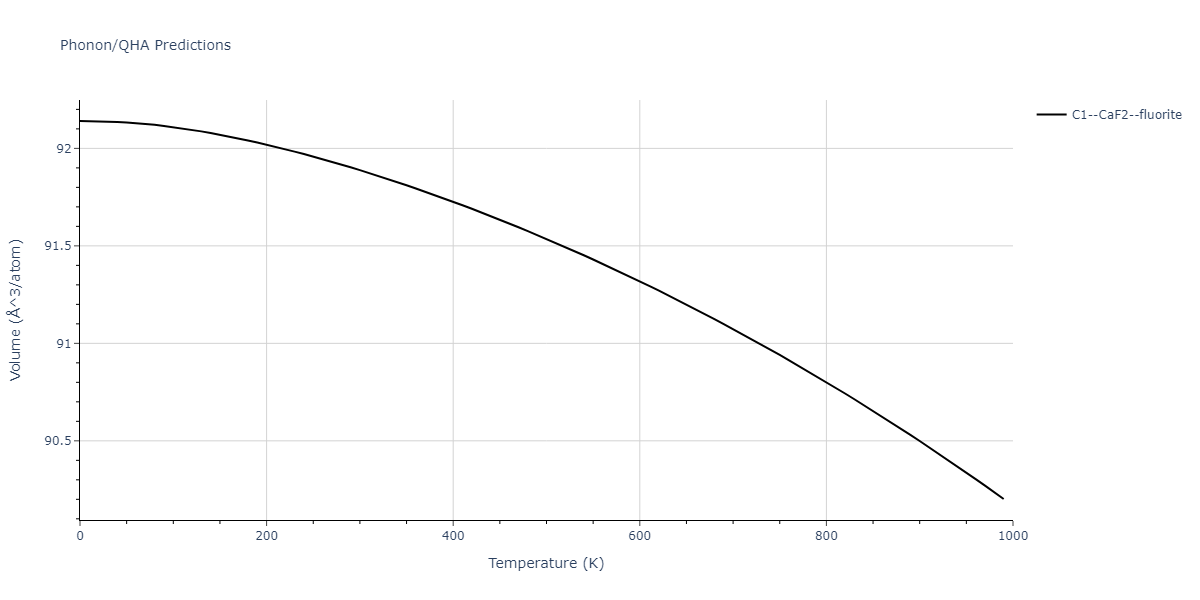
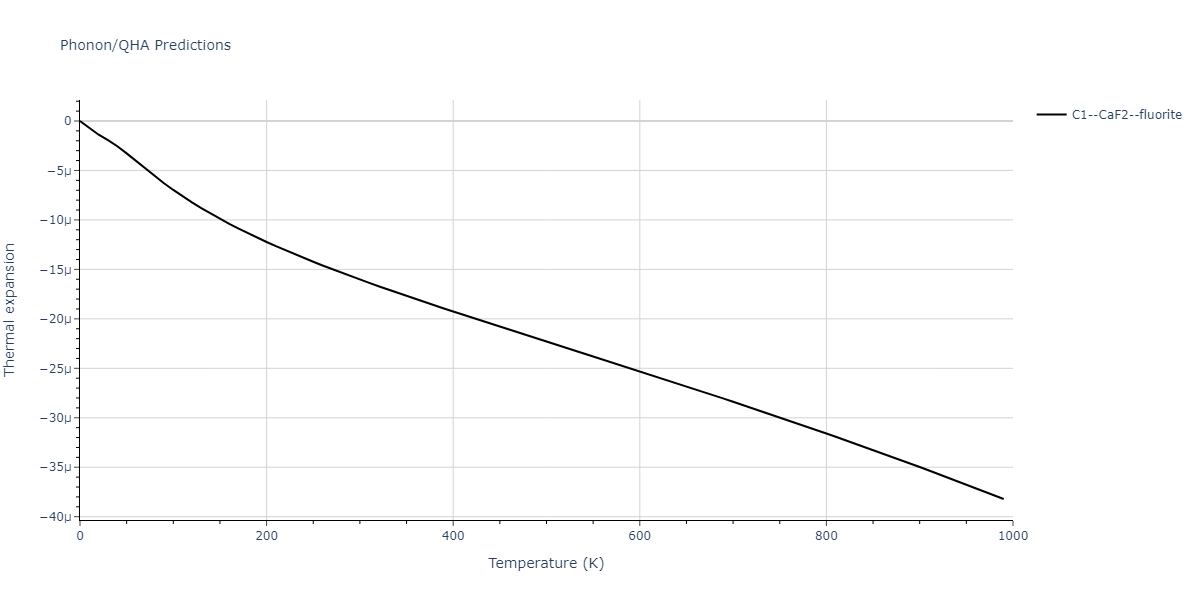
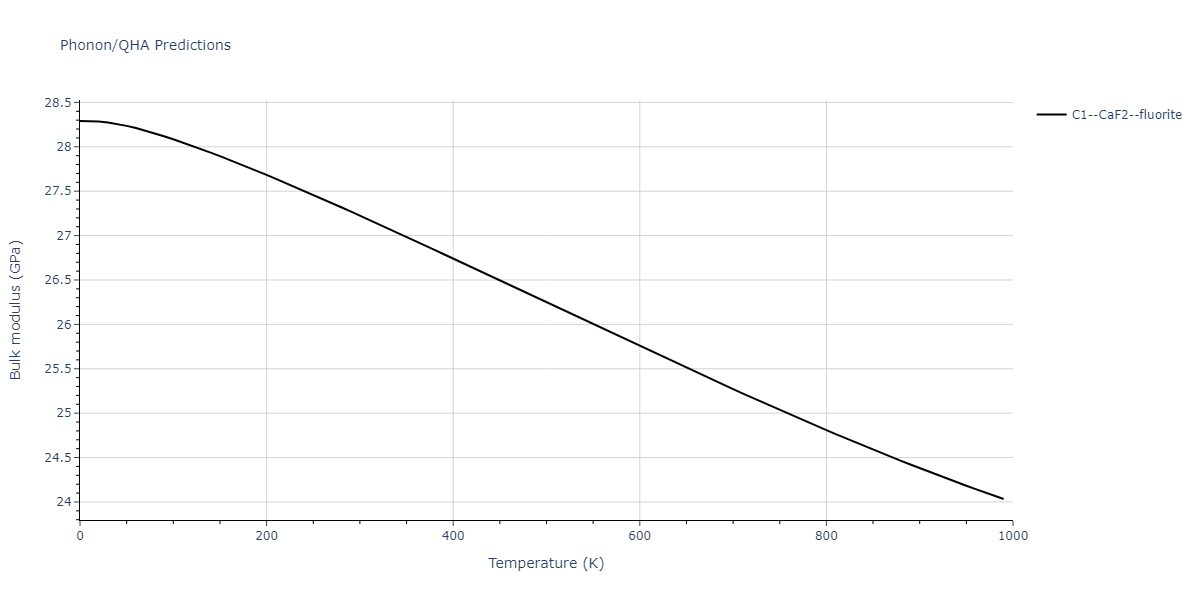
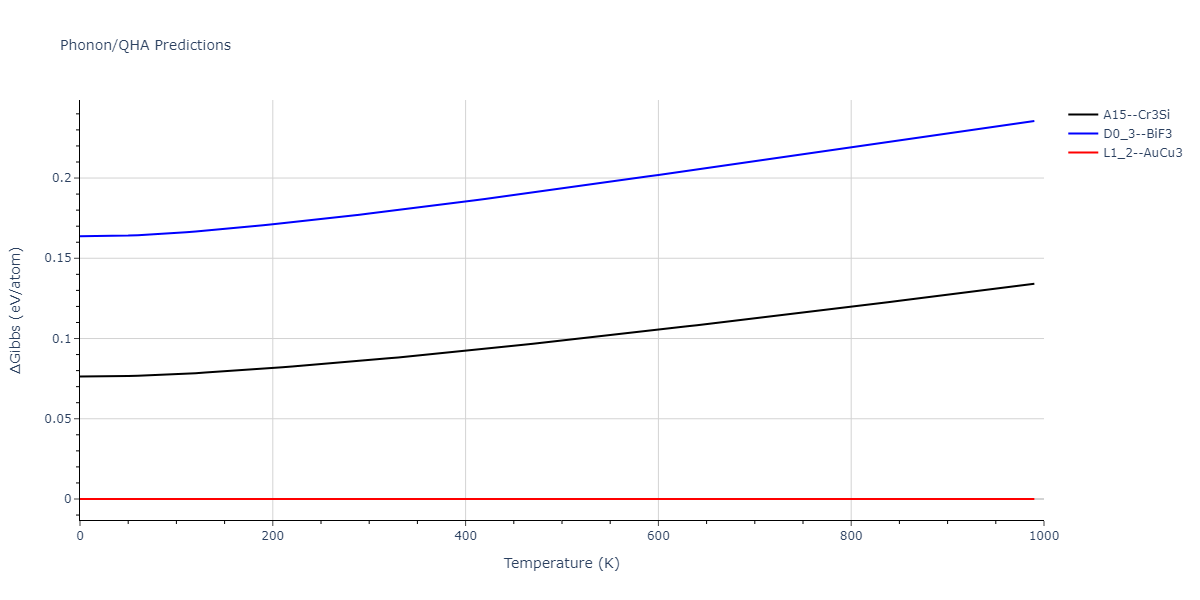
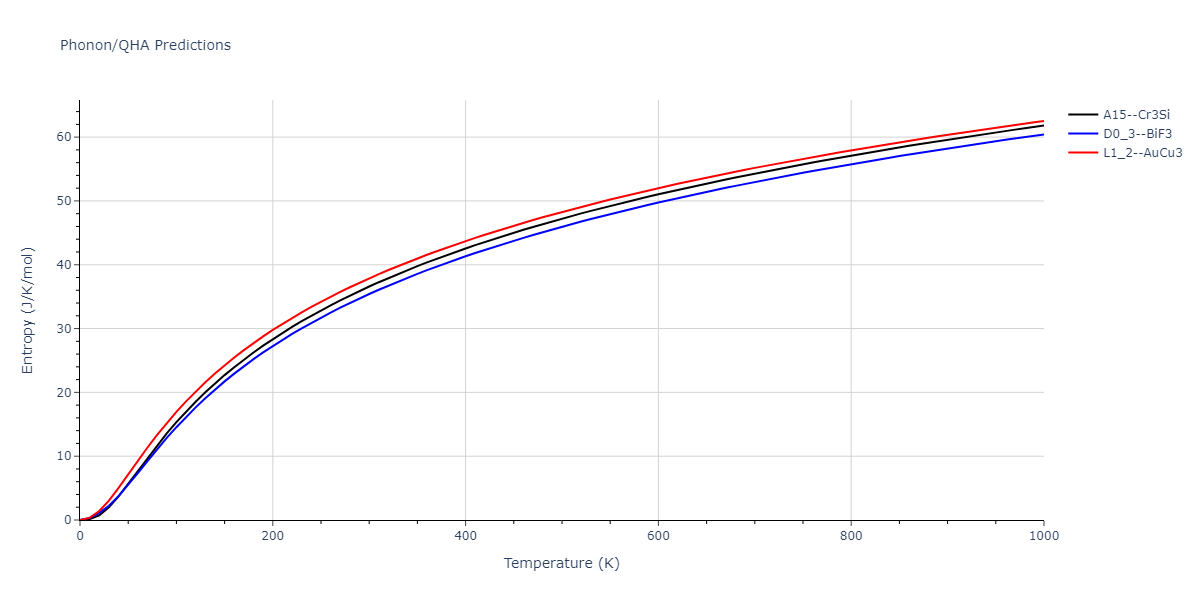
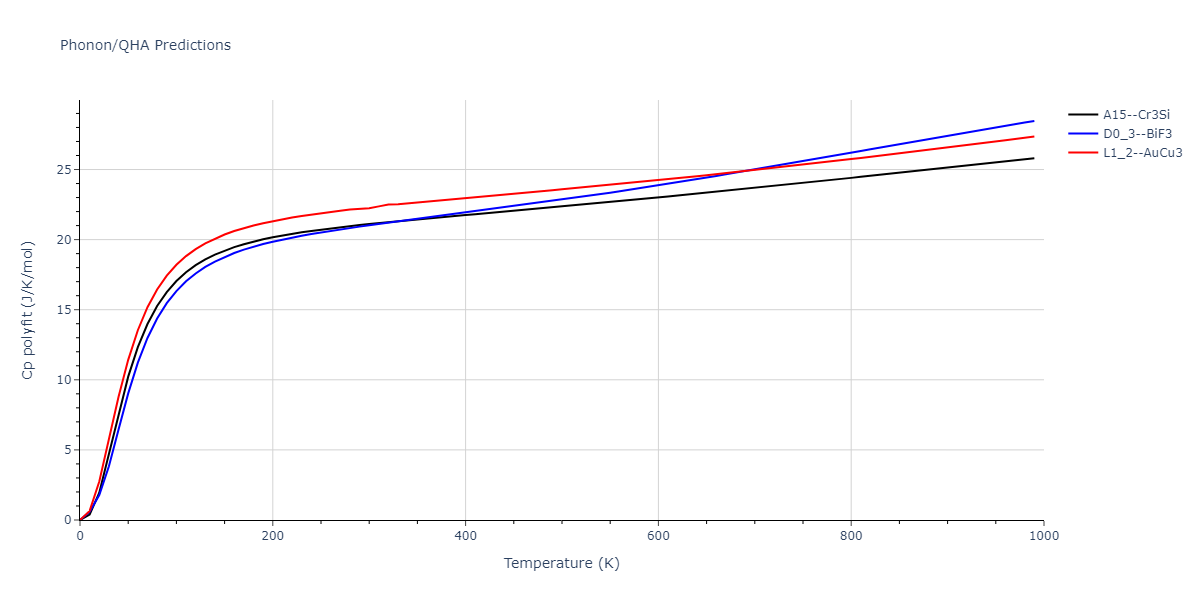
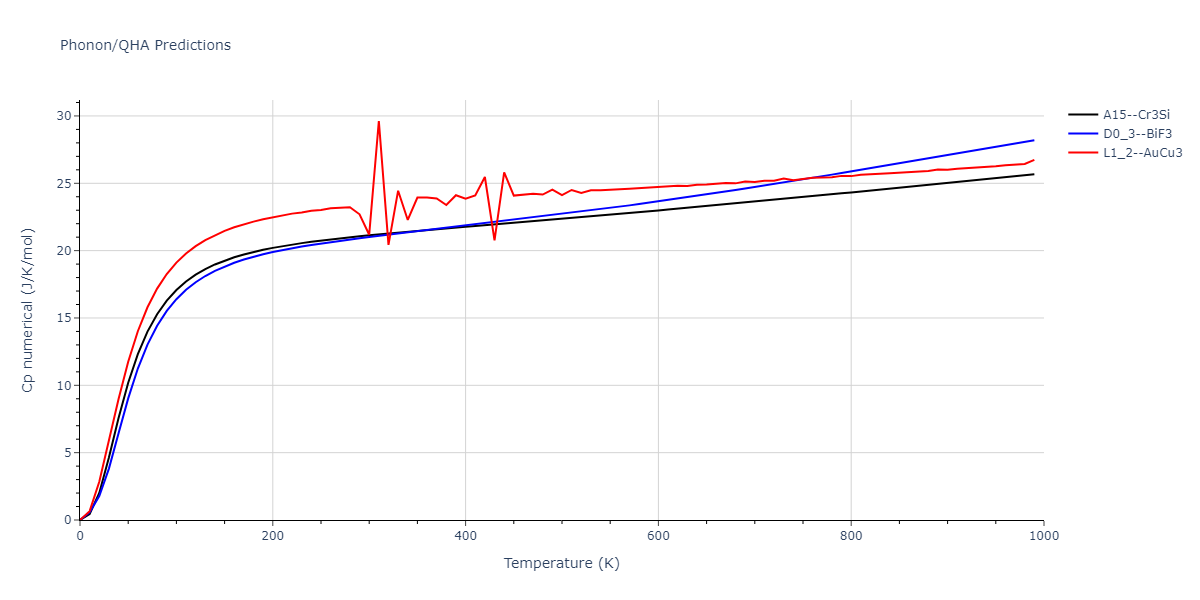
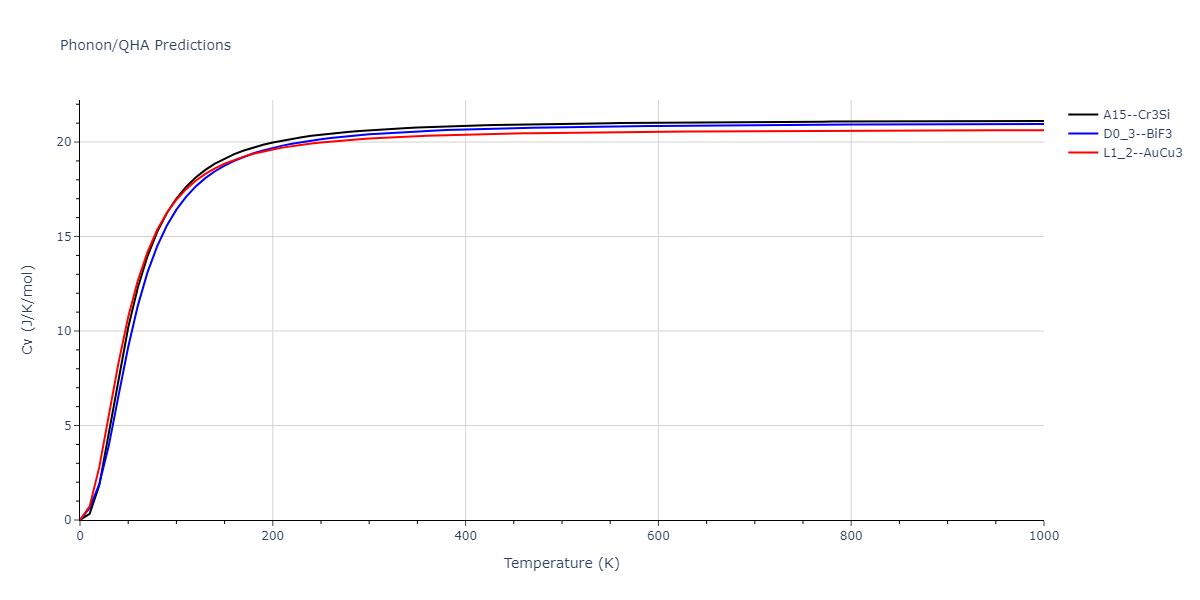
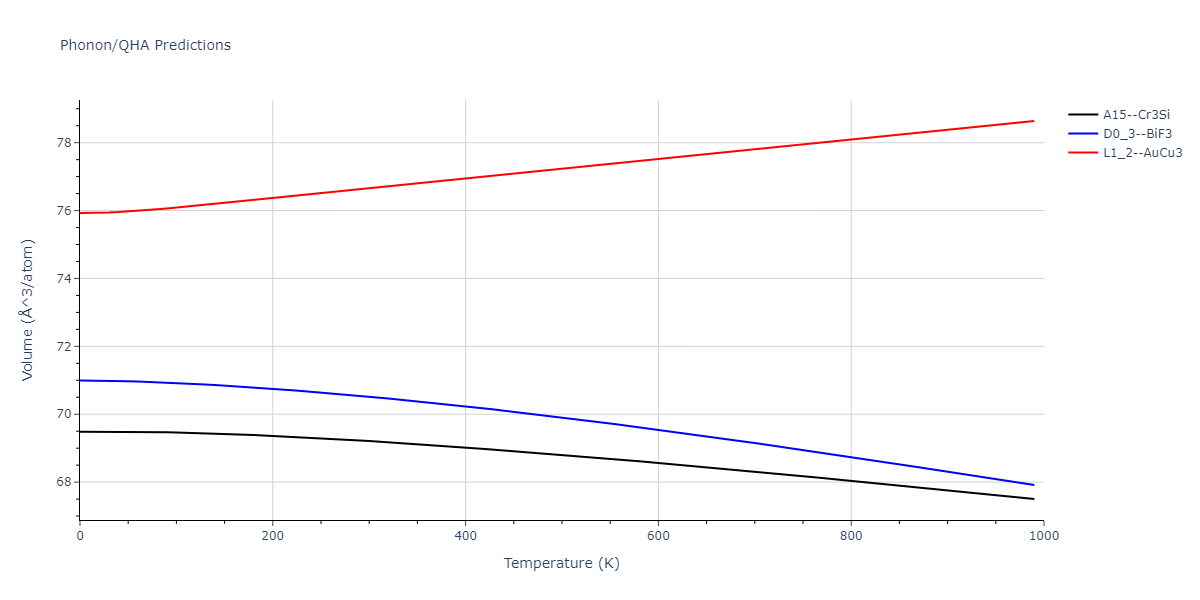
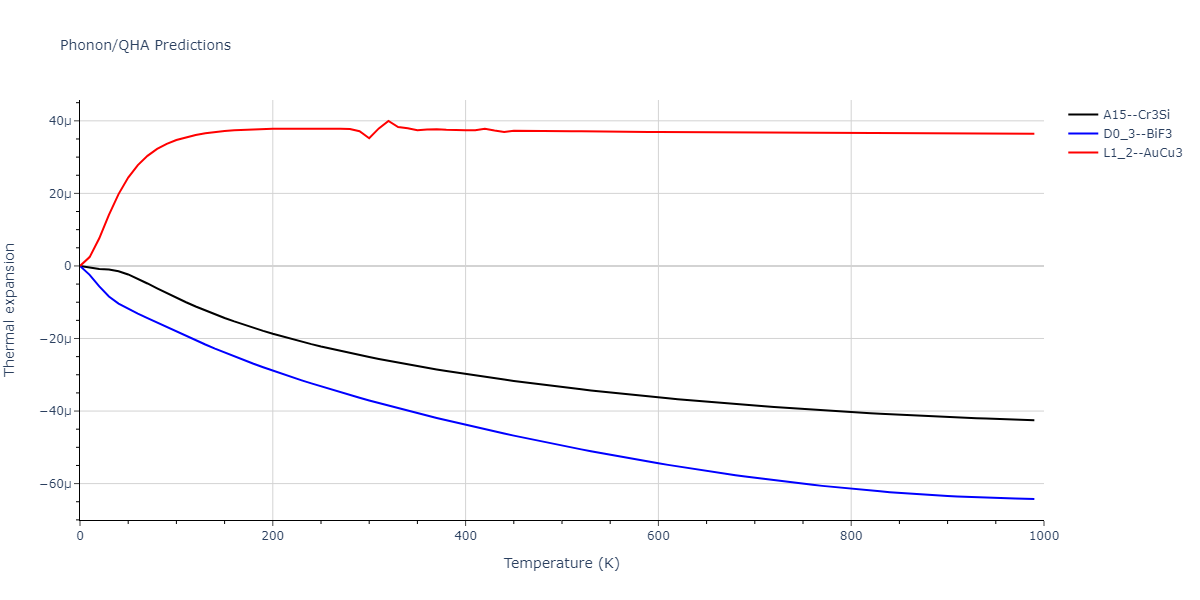
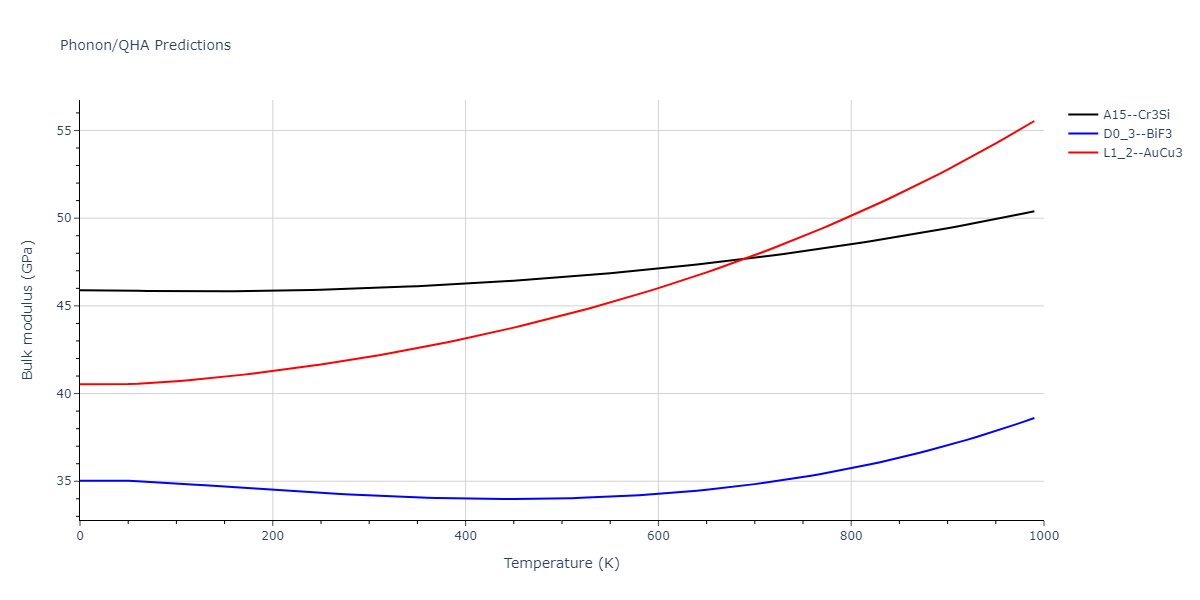
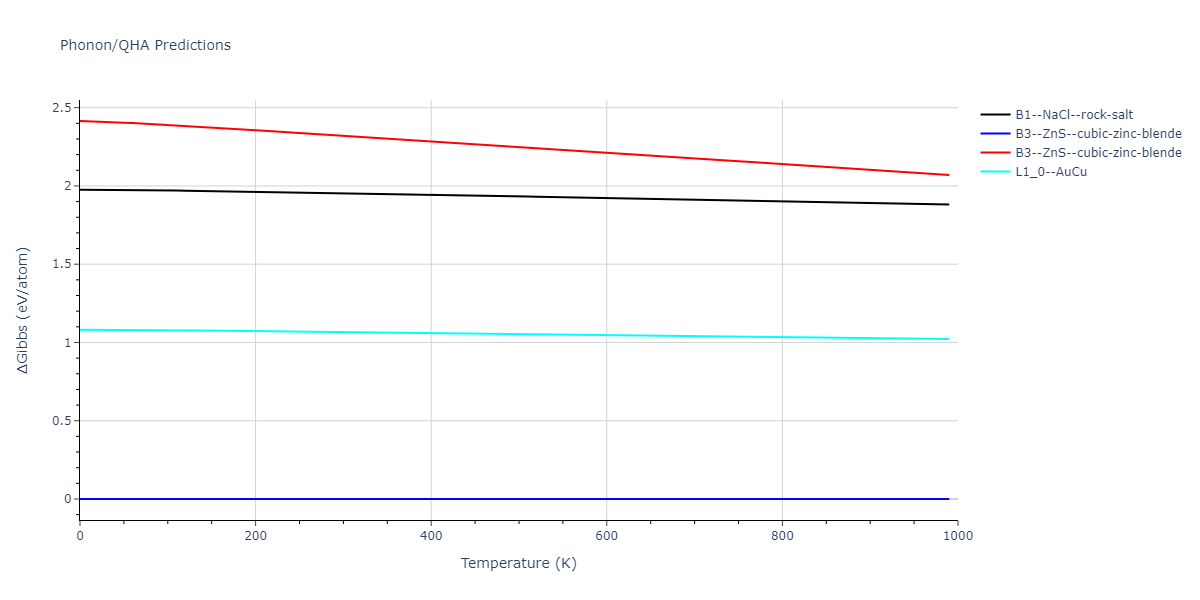
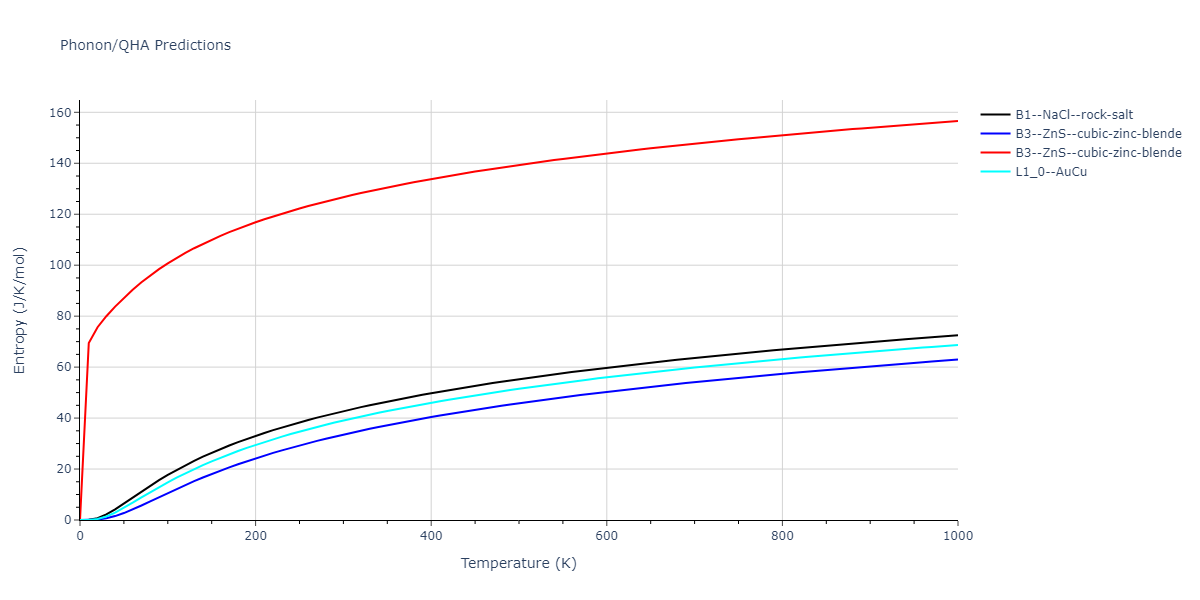
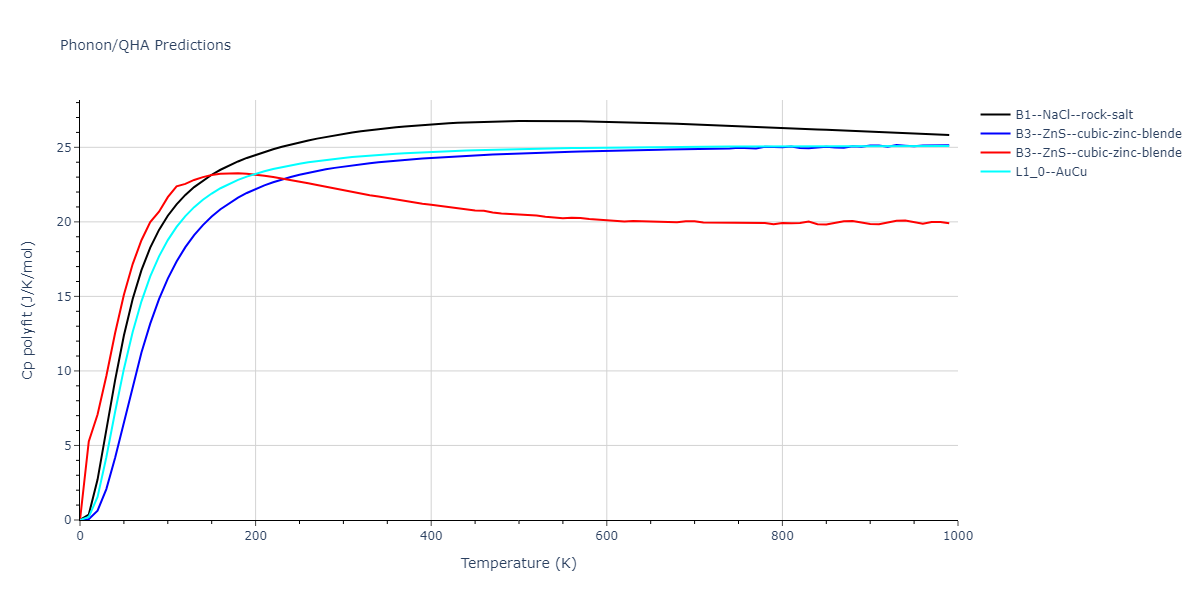
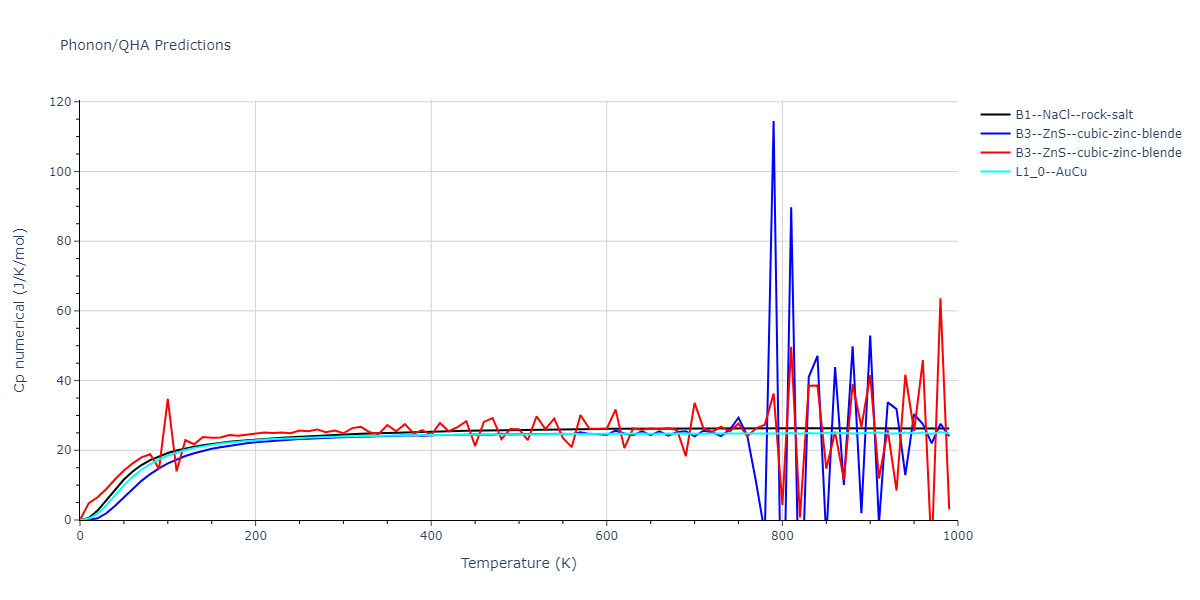
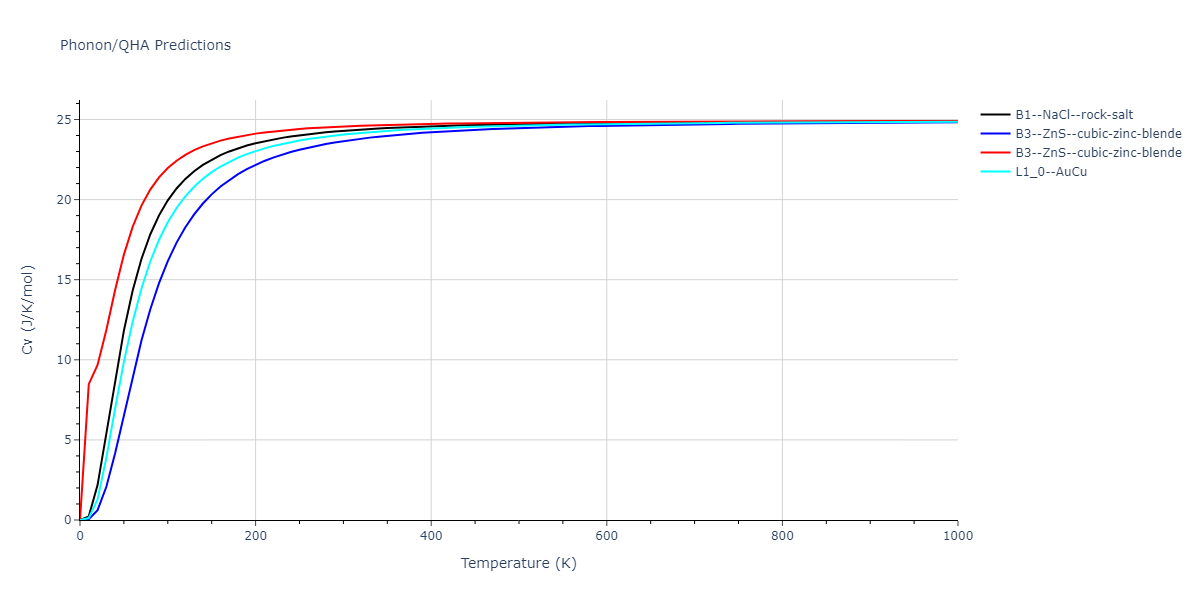
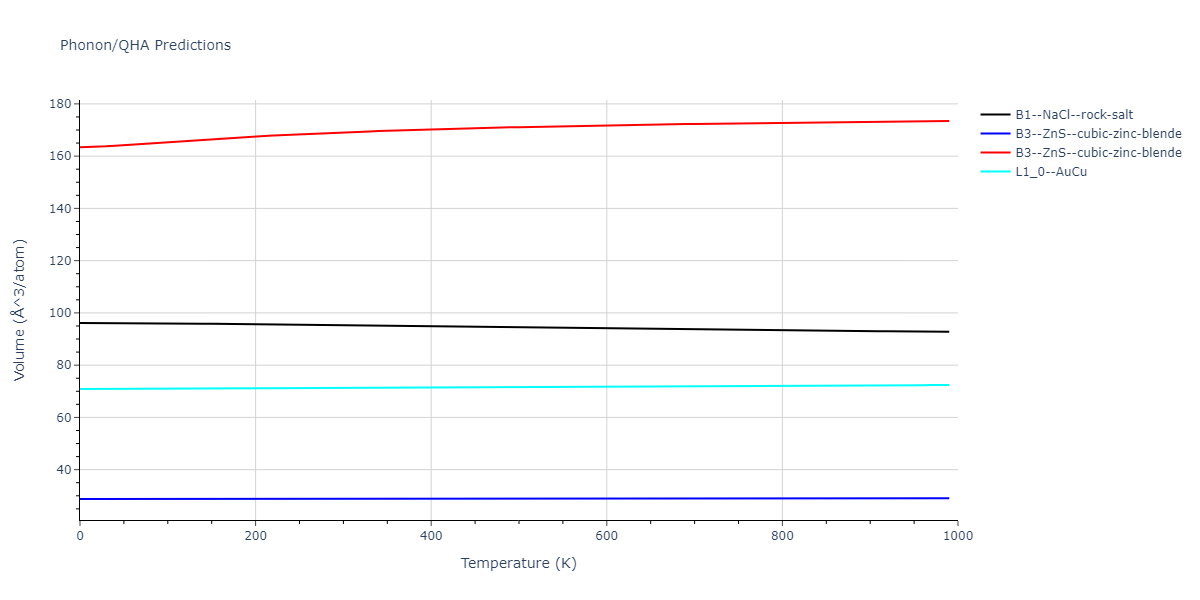
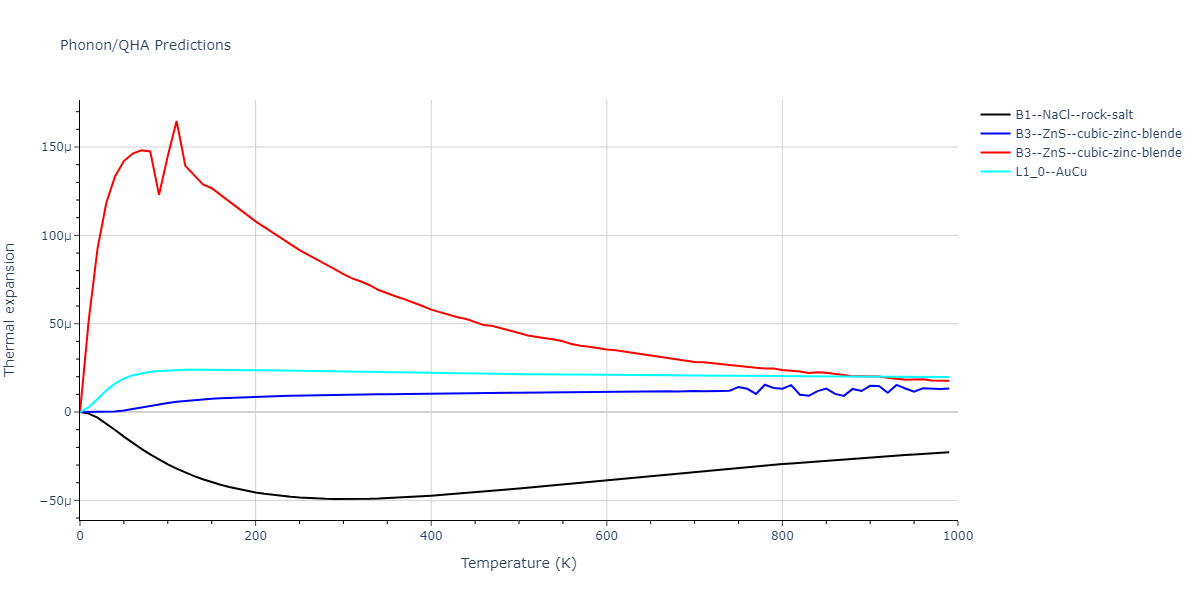
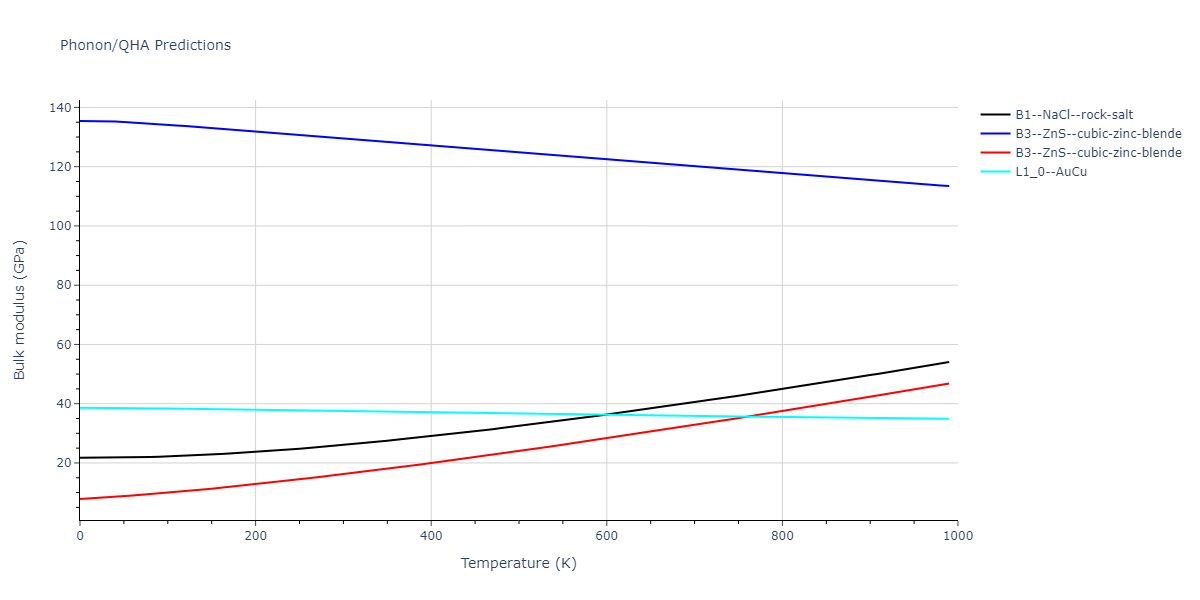
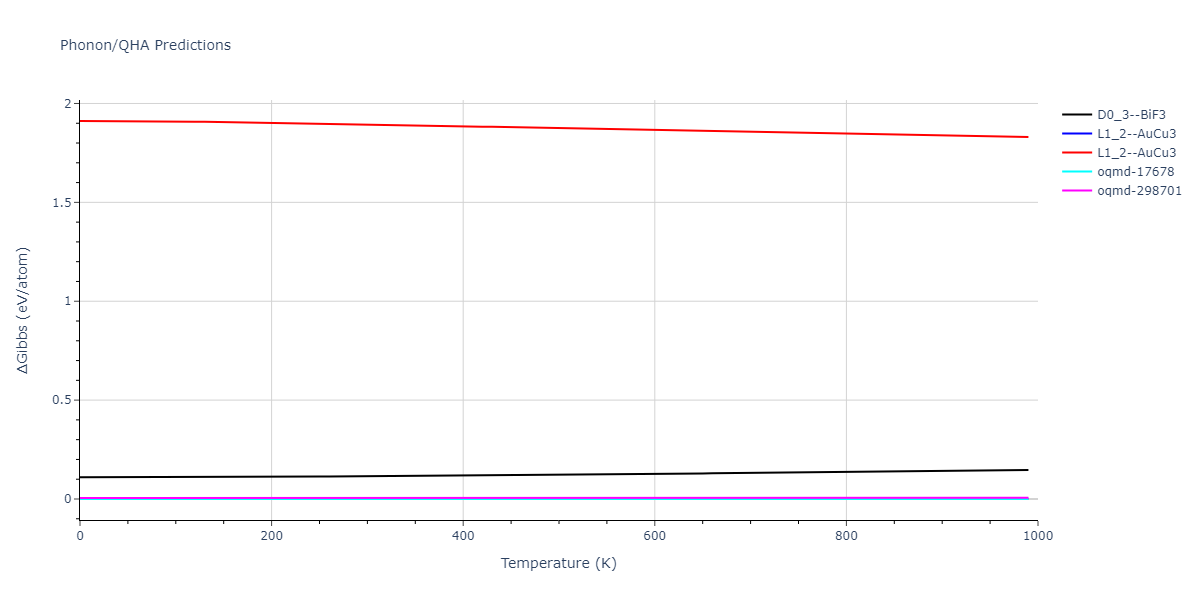
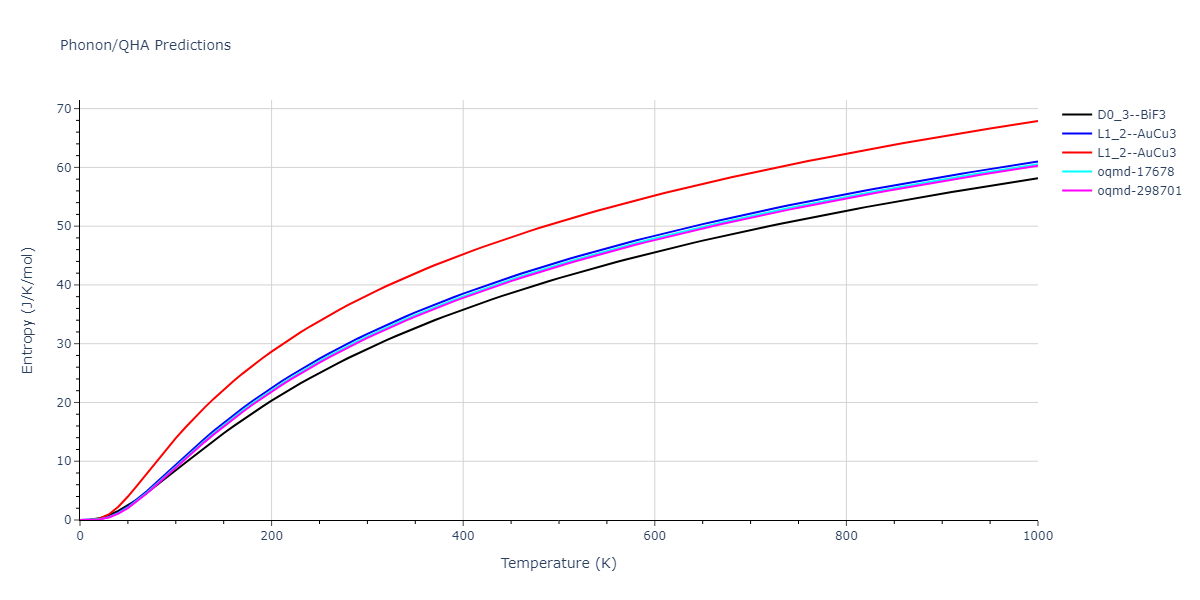
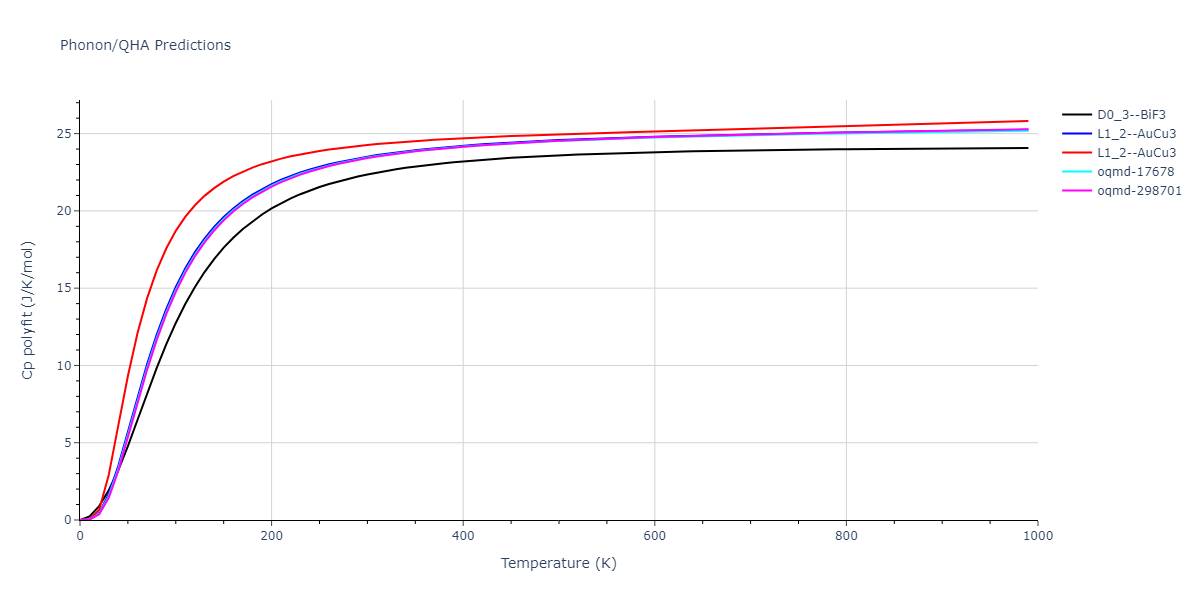
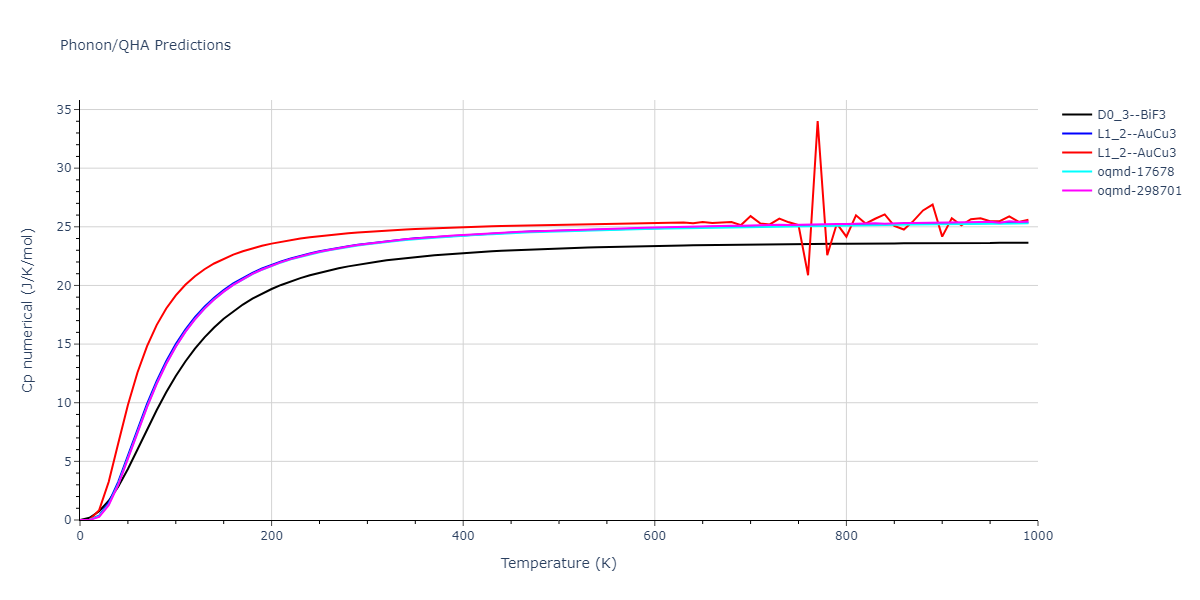
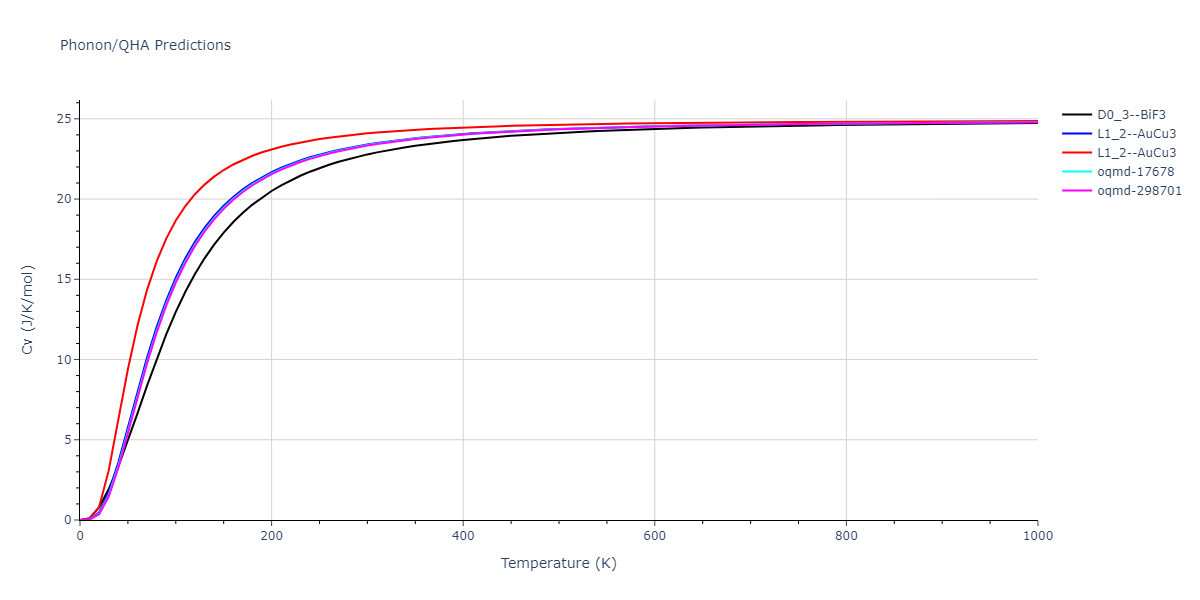
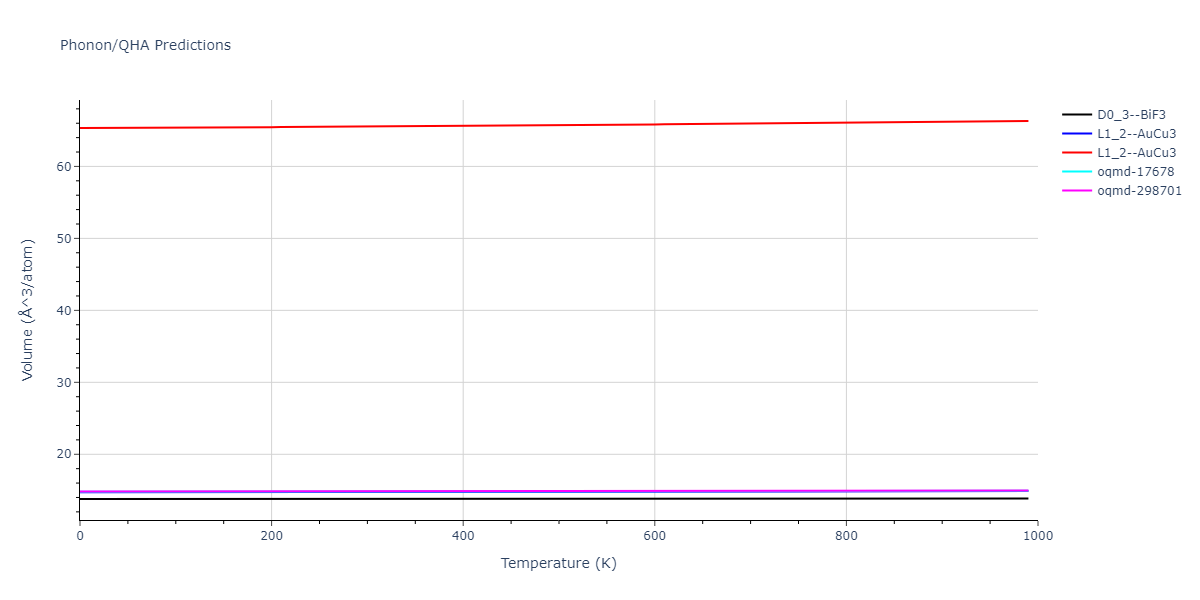
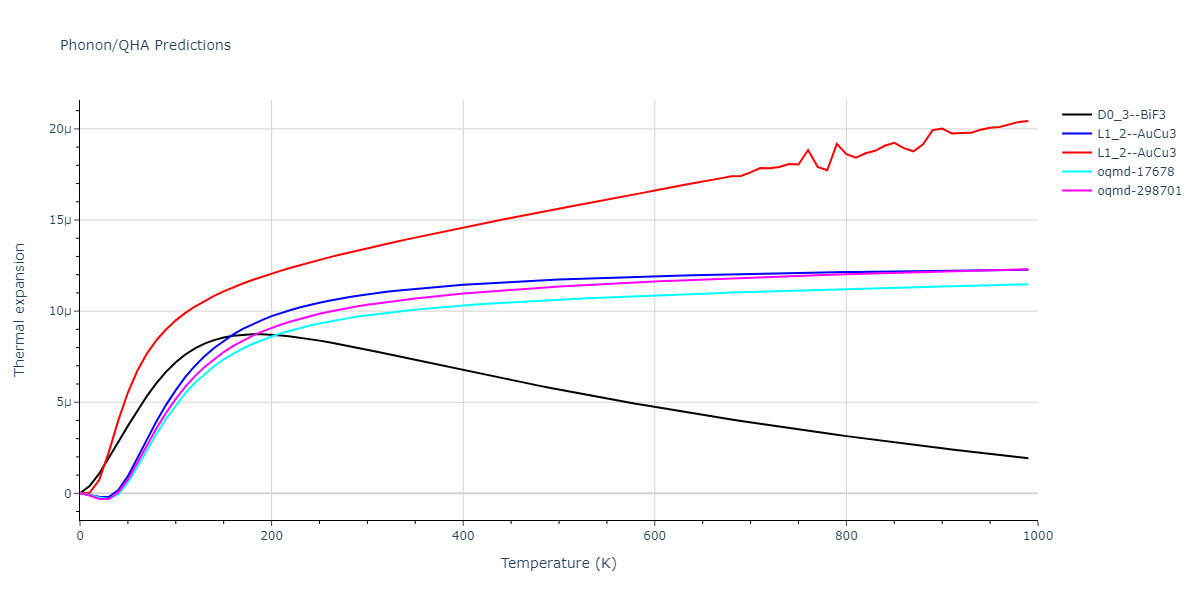
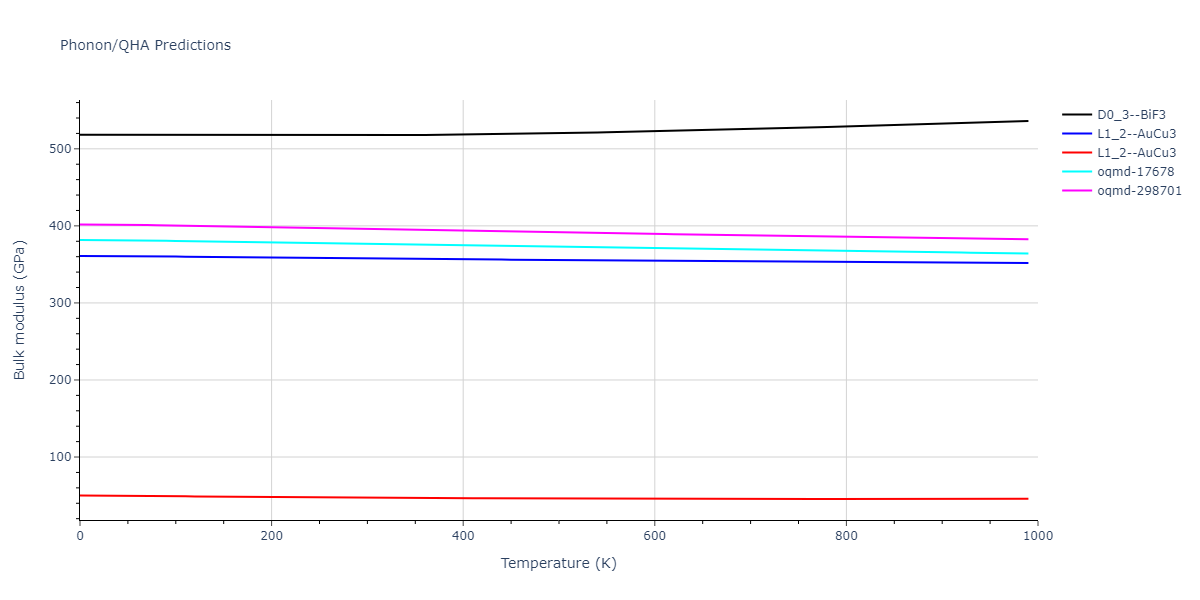
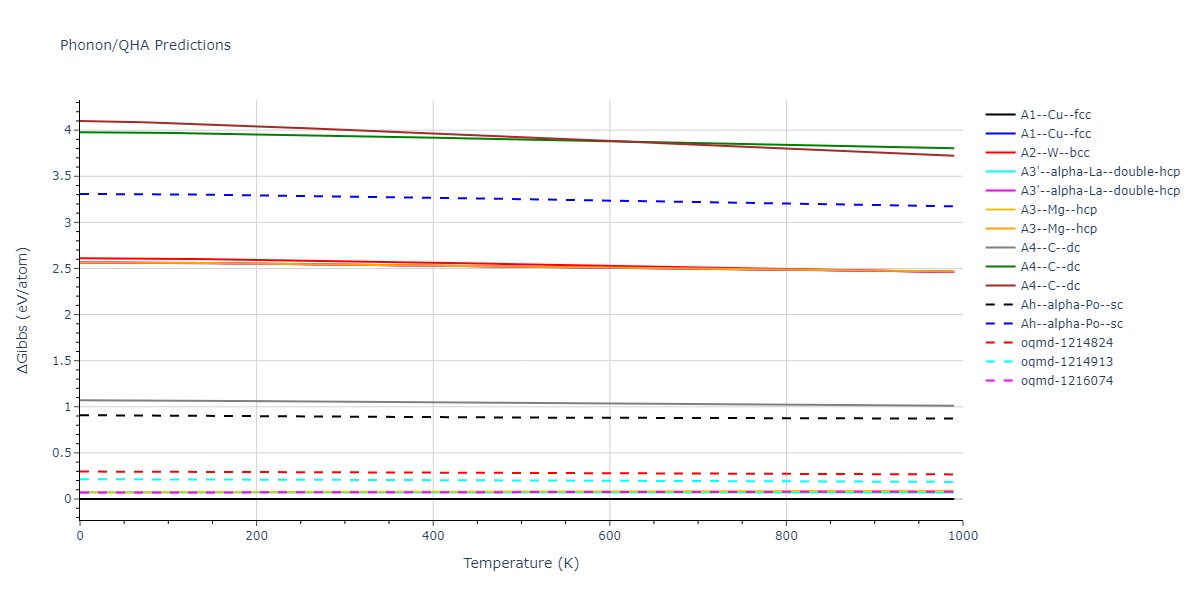
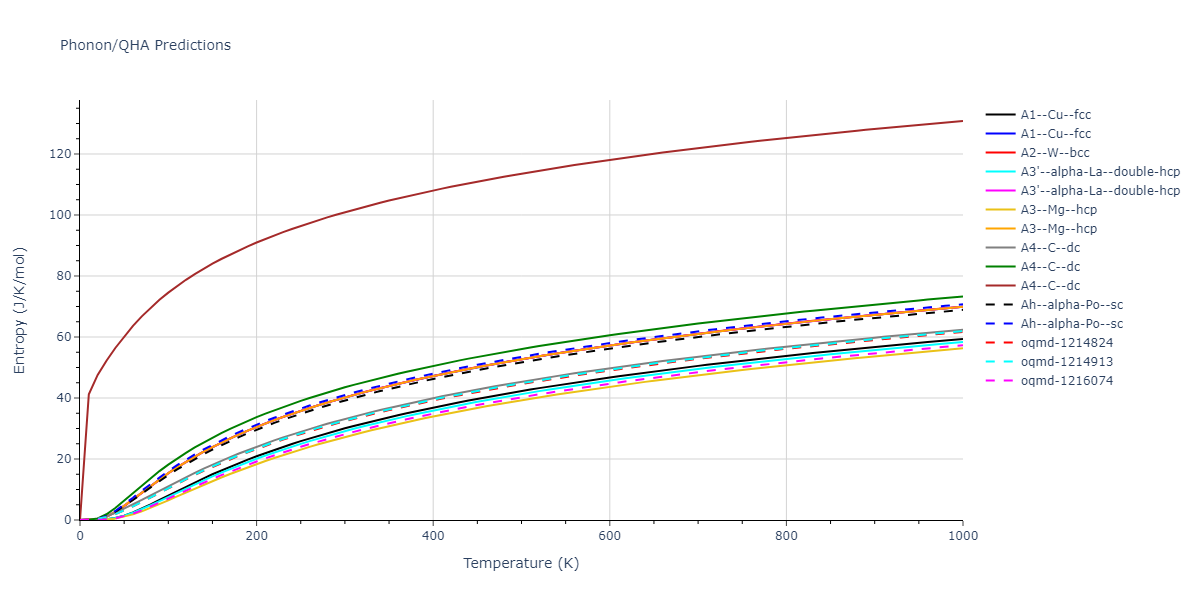
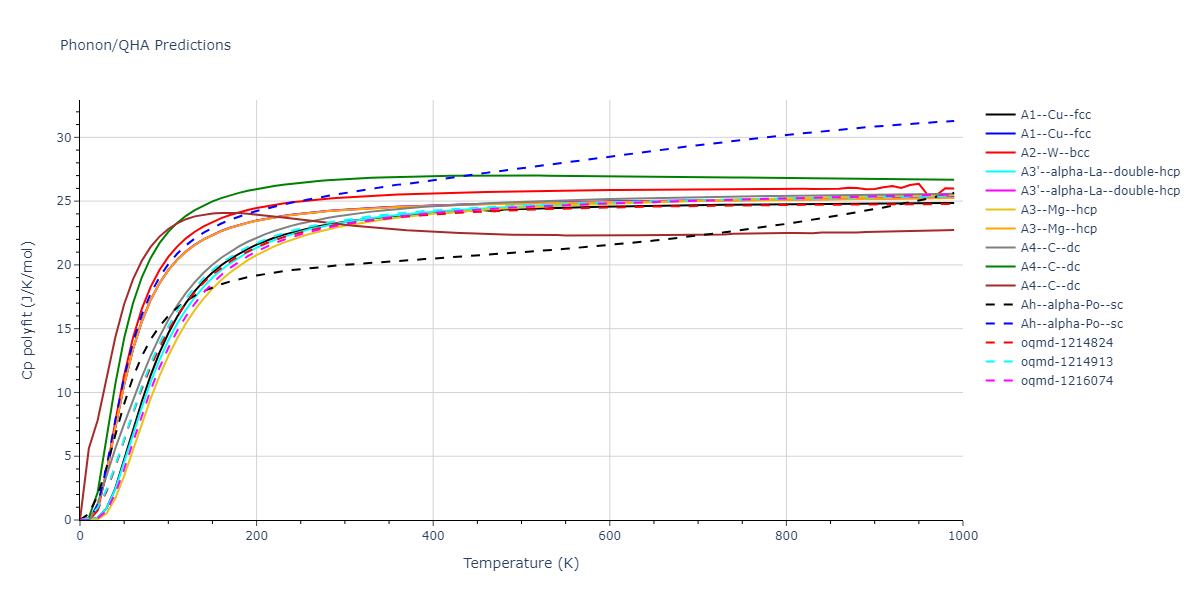
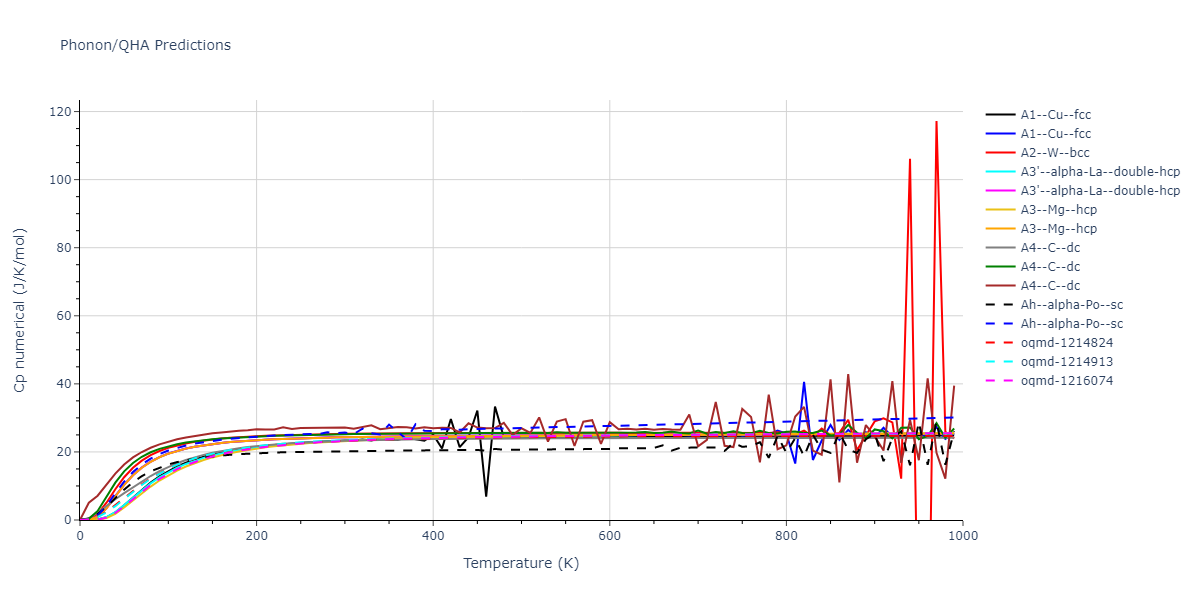
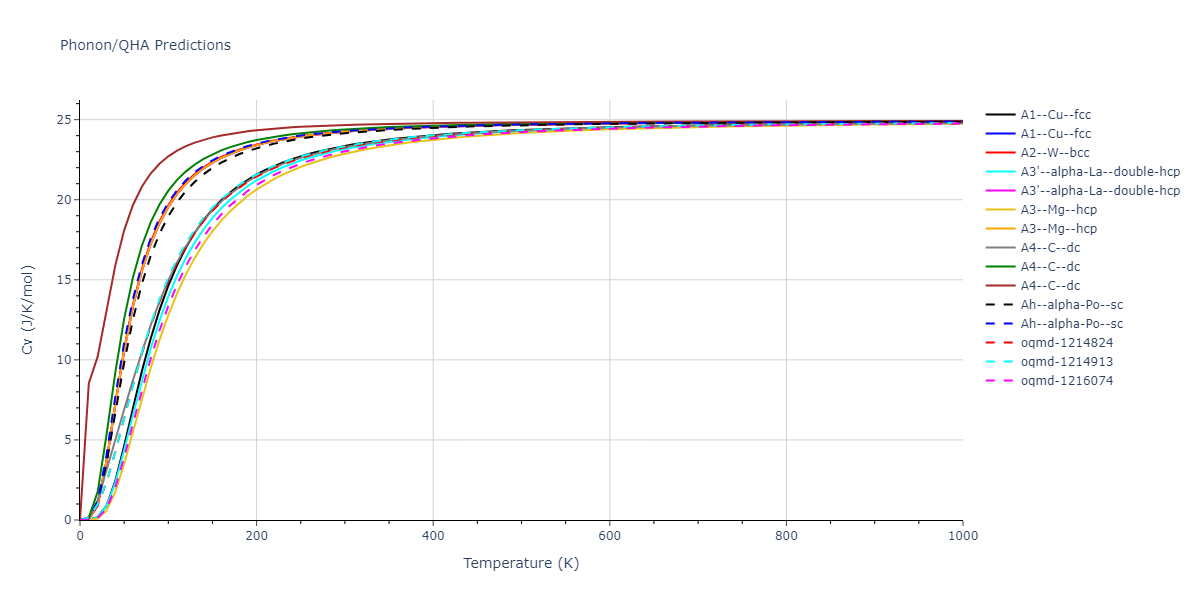
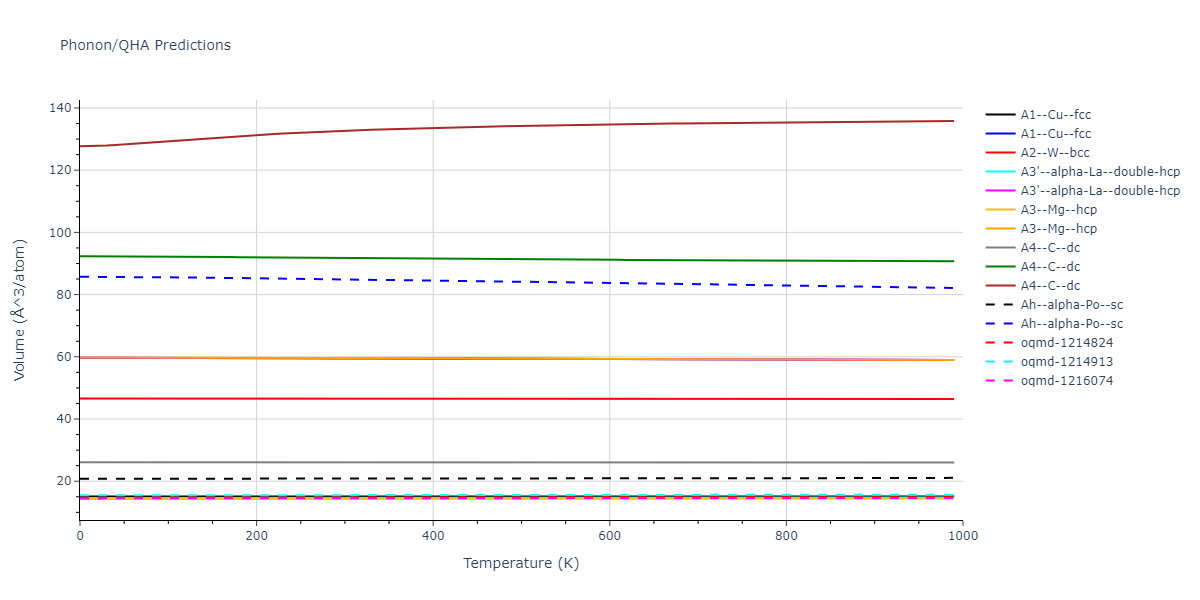
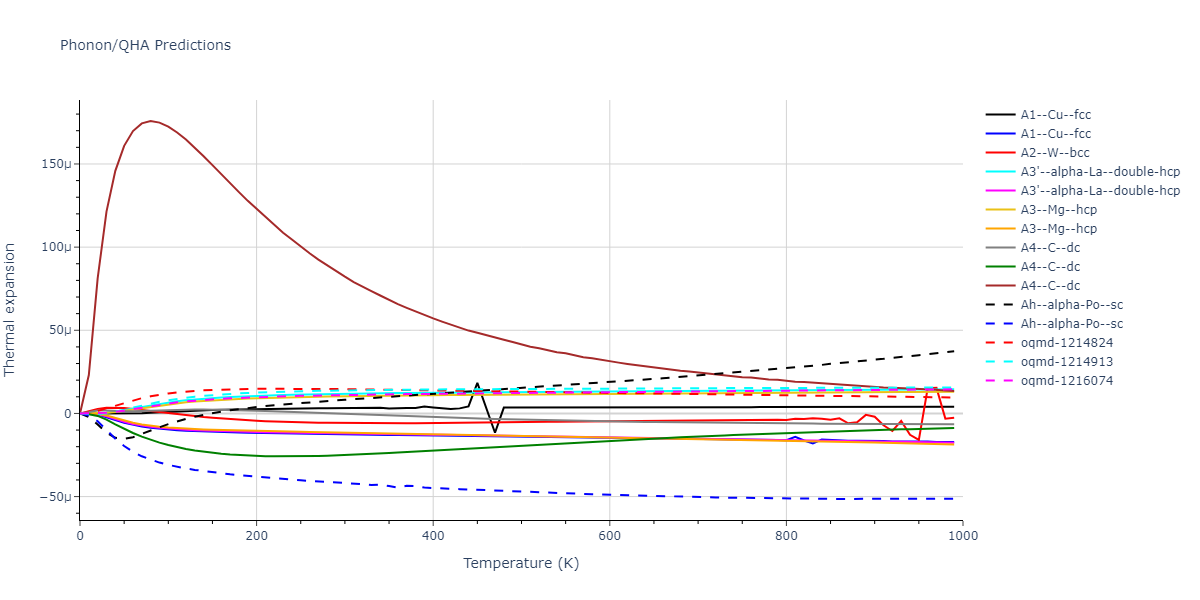
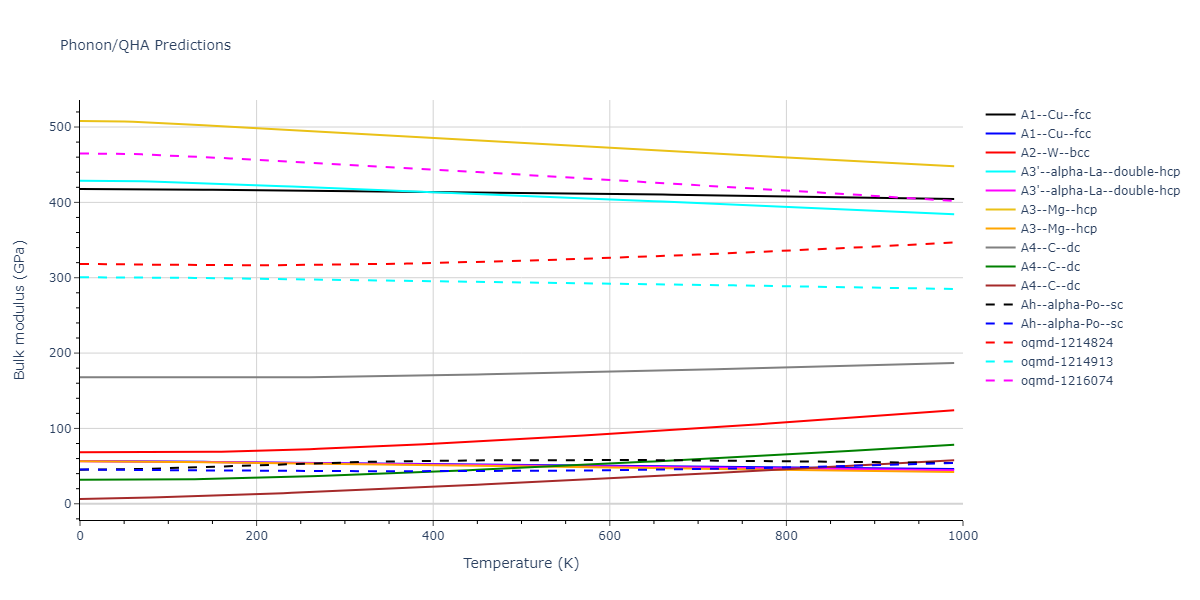
Free Surface Formation Energy Predictions
Static free surface formation energies are displayed for select crystals. The values displayed here are obtained by taking a perfect periodic bulk crystal, slicing along a crystallographic plane, and using force minimization to statically relax the surfaces. The free surface formation energy is computed by comparing the energy of the defect system to the bulk system and dividing by the total surface area created by the cut.
The calculation method used is available as the iprPy surface_energy_static calculation method.
Notes and Disclaimers:
- These values are meant to be guidelines for comparing potentials, not the absolute values for any potential's properties. Values listed here may change if the calculation methods are updated due to improvements/corrections. Variations in the values may occur for variations in calculation methods, simulation software and implementations of the interatomic potentials.
- The calculation only performs straight cuts along crystallographic planes and static relaxations. Lower energy configurations may exist that require atomic restructuring of the surfaces.
- Multiple values may be listed for a given plane followed by a number indicating different unique atomic planar cuts for the same theoretical plane.
- NIST disclaimer
Version Information:
- 2019-11-14. Calculations for the alternate #2 plane slices are now different from the #1 plane slices.
- 2019-08-07. Data added.
| Surface | γfs (mJ/m2) |
|---|---|
| (100) | 582.59 |
| (111) | 600.64 |
| (110) | 621.97 |
| (210) | 630.97 |
| (310) | 631.41 |
| (332) | 631.49 |
| (322) | 634.94 |
| (320) | 635.14 |
| (311) | 638.72 |
| (221) | 641.29 |
| (331) | 645.04 |
| (211) | 645.10 |
| (321) | 651.22 |
| Surface | γfs (mJ/m2) |
|---|---|
| (1012) 2 | 603.63 |
| (0001) | 612.28 |
| (1120) | 624.00 |
| (1011) 1 | 630.60 |
| (2241) | 637.80 |
| (2021) 1 | 639.22 |
| (2132) 2 | 646.06 |
| (2021) 2 | 646.53 |
| (1121) | 647.80 |
| (2130) | 648.73 |
| (2131) 1 | 650.25 |
| (2131) 2 | 651.16 |
| (1010) | 651.38 |
| (2112) | 655.46 |
| (2132) 1 | 659.91 |
| (1011) 2 | 661.05 |
| (1012) 1 | 675.38 |
| Surface | γfs (mJ/m2) |
|---|---|
| (1011) 2 | 614.20 |
| (1120) | 629.38 |
| (0001) | 629.49 |
| (1010) 1 | 632.76 |
| (1012) 1 | 634.26 |
| (2021) 1 | 644.23 |
| (2130) 1 | 651.79 |
| (2112) | 655.07 |
| (2130) 2 | 655.20 |
| (2131) 2 | 656.02 |
| (2241) | 656.51 |
| (2132) 1 | 662.63 |
| (2021) 2 | 664.24 |
| (1121) | 667.56 |
| (2132) 2 | 668.64 |
| (1010) 2 | 681.09 |
| (2131) 1 | 685.10 |
| (1012) 2 | 688.84 |
| (1011) 1 | 761.00 |
| Surface | γfs (mJ/m2) |
|---|---|
| (332) | -755.89 |
| (322) | -712.98 |
| (331) | -561.26 |
| (320) | -442.26 |
| (310) | -381.19 |
| (221) | -206.73 |
| (210) | -181.14 |
| (100) | 541.19 |
| (111) | 568.41 |
| (110) | 575.06 |
| (211) | 608.04 |
| (311) | 611.59 |
| (321) | 616.24 |
| Surface | γfs (mJ/m2) |
|---|---|
| (111) | 501.34 |
| (332) | 556.42 |
| (322) | 562.79 |
| (221) | 580.51 |
| (100) | 581.38 |
| (211) | 592.81 |
| (331) | 599.86 |
| (311) | 613.83 |
| (110) | 617.44 |
| (321) | 621.74 |
| (310) | 644.52 |
| (320) | 646.22 |
| (210) | 651.49 |
| Surface | γfs (mJ/m2) |
|---|---|
| (332) 1 | -11163.33 |
| (332) 2 | -9986.89 |
| (331) | -9358.93 |
| (322) | -9084.24 |
| (321) 1 | -8193.33 |
| (321) 2 | -7034.66 |
| (221) | -5911.87 |
| (320) | -5521.40 |
| (311) | -4846.12 |
| (310) | -4692.07 |
| (211) 2 | -3374.52 |
| (211) 1 | -2410.07 |
| (210) | -1906.25 |
| (111) | 2002.83 |
| (100) | 2274.11 |
| (110) | 5245.94 |
| Surface | γfs (mJ/m2) |
|---|---|
| (211) 2 | 488.25 |
| (332) 2 | 519.67 |
| (210) | 523.31 |
| (311) | 528.36 |
| (321) 2 | 532.05 |
| (321) 1 | 532.43 |
| (111) | 536.83 |
| (221) | 537.16 |
| (322) | 537.67 |
| (331) | 548.39 |
| (100) | 553.05 |
| (310) | 553.84 |
| (332) 1 | 557.57 |
| (320) | 558.01 |
| (211) 1 | 573.27 |
| (110) | 573.66 |
| Surface | γfs (mJ/m2) |
|---|---|
| (110) | 5547.96 |
| (211) | 5631.61 |
| (321) | 5639.92 |
| (320) | 5651.70 |
| (221) | 5681.86 |
| (331) | 5689.90 |
| (310) | 5700.94 |
| (332) | 5724.84 |
| (210) | 5732.39 |
| (322) | 5747.02 |
| (311) | 5823.23 |
| (111) | 5869.04 |
| (100) | 6481.34 |
| Surface | γfs (mJ/m2) |
|---|---|
| (332) | -1371.57 |
| (321) | -1189.45 |
| (320) | -80.68 |
| (311) | 22.04 |
| (211) | 38.03 |
| (310) | 43.09 |
| (210) | 151.56 |
| (110) | 540.52 |
| (100) | 566.24 |
| (331) | 598.68 |
| (221) | 617.62 |
| (111) | 643.28 |
| Surface | γfs (mJ/m2) |
|---|---|
| (111) 2 | 65.49 |
| (331) 2 | 78.07 |
| (110) | 80.21 |
| (321) | 90.92 |
| (211) | 92.57 |
| (320) | 94.35 |
| (332) | 96.56 |
| (210) | 101.41 |
| (311) 1 | 102.54 |
| (311) 2 | 102.54 |
| (310) | 107.54 |
| (322) | 109.82 |
| (221) | 113.16 |
| (100) | 113.32 |
| (331) 1 | 129.74 |
| (111) 1 | 195.52 |
| Surface | γfs (mJ/m2) |
|---|---|
| (101) | 5378.51 |
| (001) | 5466.32 |
| (201) | 5535.13 |
| (100) | 5543.67 |
| (211) | 5570.61 |
| (111) | 5753.14 |
| (112) | 5874.83 |
| (110) | 6173.67 |
| Surface | γfs (mJ/m2) |
|---|---|
| (210) | -28065.72 |
| (221) | -24037.75 |
| (331) | -22484.10 |
| (321) | -21950.57 |
| (332) | -13487.32 |
| (322) | -11169.88 |
| (311) | -9623.23 |
| (211) | -5080.99 |
| (320) | -3181.86 |
| (100) | 150.17 |
| (310) | 190.17 |
| (110) | 212.88 |
| (111) | 261.15 |
| Surface | γfs (mJ/m2) |
|---|---|
| (111) | 2797.53 |
| (100) | 2825.95 |
| (311) | 2829.59 |
| (210) | 2851.45 |
| (110) | 2872.83 |
| (310) | 2879.21 |
| (332) | 2893.37 |
| (320) | 2903.07 |
| (321) | 2903.13 |
| (322) | 2909.22 |
| (331) | 2913.85 |
| (211) | 2915.57 |
| (221) | 2916.95 |
| Surface | γfs (mJ/m2) |
|---|---|
| (111) | 520.80 |
| (100) | 555.88 |
| (332) | 569.66 |
| (322) | 575.23 |
| (221) | 590.03 |
| (211) | 600.03 |
| (331) | 605.36 |
| (110) | 613.21 |
| (311) | 614.41 |
| (321) | 626.78 |
| (310) | 629.97 |
| (320) | 638.71 |
| (210) | 642.06 |
| Surface | γfs (mJ/m2) |
|---|---|
| (331) | -59063.36 |
| (322) | -56115.98 |
| (221) | -39782.07 |
| (210) | -28595.37 |
| (111) | -22328.25 |
| (100) | -4810.96 |
| (310) | 699.58 |
| (110) | 712.56 |
| (320) | 724.61 |
| (311) | 759.33 |
| (321) | 784.27 |
| (211) | 788.85 |
| (332) | 799.89 |
| Surface | γfs (mJ/m2) |
|---|---|
| (0001) | 2689.61 |
| (1012) 2 | 2739.83 |
| (1120) | 2741.05 |
| (1011) 1 | 2756.68 |
| (2241) | 2780.39 |
| (2130) | 2794.02 |
| (2021) 1 | 2797.75 |
| (2132) 2 | 2799.89 |
| (1121) | 2801.34 |
| (2131) 1 | 2801.77 |
| (2112) | 2804.79 |
| (2021) 2 | 2814.60 |
| (2131) 2 | 2816.81 |
| (1010) | 2821.94 |
| (2132) 1 | 2831.53 |
| (1012) 1 | 2904.18 |
| (1011) 2 | 2976.11 |
| Surface | γfs (mJ/m2) |
|---|---|
| (0001) | 520.80 |
| (1012) 2 | 544.77 |
| (2021) 1 | 565.79 |
| (1011) 1 | 567.66 |
| (2021) 2 | 600.72 |
| (1120) | 612.83 |
| (2131) 1 | 621.56 |
| (1010) | 624.67 |
| (2132) 2 | 625.11 |
| (2241) | 625.17 |
| (2132) 1 | 630.58 |
| (1011) 2 | 632.23 |
| (1012) 1 | 633.66 |
| (1121) | 633.79 |
| (2130) | 639.18 |
| (2112) | 640.15 |
| (2131) 2 | 648.80 |
| Surface | γfs (mJ/m2) |
|---|---|
| (1010) 1 | 2738.64 |
| (0001) | 2761.88 |
| (1011) 2 | 2770.51 |
| (1120) | 2797.19 |
| (2112) | 2818.22 |
| (2130) 1 | 2834.11 |
| (2021) 1 | 2848.06 |
| (1012) 1 | 2852.36 |
| (2241) | 2853.22 |
| (2131) 2 | 2859.91 |
| (1121) | 2865.68 |
| (2130) 2 | 2869.16 |
| (1012) 2 | 2879.98 |
| (2132) 1 | 2884.41 |
| (2131) 1 | 2900.47 |
| (2132) 2 | 2908.79 |
| (2021) 2 | 2973.67 |
| (1010) 2 | 3071.91 |
| (1011) 1 | 3083.14 |
| Surface | γfs (mJ/m2) |
|---|---|
| (0001) | 520.80 |
| (1011) 2 | 544.96 |
| (1010) 1 | 556.09 |
| (2021) 1 | 566.85 |
| (1012) 1 | 597.79 |
| (2130) 1 | 611.65 |
| (1120) | 612.77 |
| (2112) | 614.59 |
| (2132) 2 | 623.48 |
| (2132) 1 | 624.49 |
| (2131) 2 | 625.11 |
| (2241) | 633.76 |
| (2021) 2 | 633.89 |
| (2131) 1 | 636.24 |
| (1121) | 640.20 |
| (2130) 2 | 667.24 |
| (1010) 2 | 694.75 |
| (1011) 1 | 724.74 |
| Surface | γfs (mJ/m2) |
|---|---|
| (111) 2 | 1300.73 |
| (331) 2 | 1454.40 |
| (110) | 1456.36 |
| (211) | 1457.47 |
| (332) | 1460.25 |
| (321) | 1494.56 |
| (311) 1 | 1506.32 |
| (322) | 1507.70 |
| (320) | 1533.65 |
| (221) | 1534.09 |
| (100) | 1545.67 |
| (310) | 1552.56 |
| (210) | 1555.25 |
| (311) 2 | 1598.32 |
| (331) 1 | 1604.61 |
| (111) 1 | 1808.60 |
| Surface | γfs (mJ/m2) |
|---|---|
| (100) | -20043.92 |
| (331) 2 | -19241.83 |
| (331) 1 | -18486.36 |
| (320) | -15865.36 |
| (311) 2 | -13866.86 |
| (210) | -9646.33 |
| (111) 2 | -6965.80 |
| (111) 1 | -6755.44 |
| (110) | -6052.06 |
| (310) | -343.92 |
| Surface | γfs (mJ/m2) |
|---|---|
| (111) 2 | 76.08 |
| (331) 2 | 90.70 |
| (110) | 93.18 |
| (321) | 108.71 |
| (211) | 112.26 |
| (320) | 112.82 |
| (332) | 120.88 |
| (210) | 122.98 |
| (311) 1 | 126.09 |
| (311) 2 | 126.09 |
| (310) | 132.25 |
| (322) | 140.29 |
| (100) | 143.21 |
| (221) | 145.07 |
| (331) 1 | 169.46 |
| (111) 1 | 274.30 |
| Surface | γfs (mJ/m2) |
|---|---|
| (332) | -20050.91 |
| (321) | -16957.27 |
| (320) | -14625.31 |
| (310) | -12457.43 |
| (322) | -11809.02 |
| (331) | -10929.75 |
| (210) | -7868.95 |
| (221) | -7615.97 |
| (311) | -7094.00 |
| (211) | -5492.20 |
| (111) | -4065.25 |
| (100) | 1813.57 |
| (110) | 1942.06 |
| Surface | γfs (mJ/m2) |
|---|---|
| (100) | 255.10 |
| (310) | 309.25 |
| (210) | 323.31 |
| (110) | 329.53 |
| (320) | 329.75 |
| (311) | 341.83 |
| (331) | 355.45 |
| (111) | 355.88 |
| (211) | 358.50 |
| (321) | 359.37 |
| (221) | 361.19 |
| (332) | 362.39 |
| (322) | 362.43 |
Point Defect Formation Energy Predictions
Static point defect formation energies, Ef, and elastic dipole tensors, pij, are displayed for select crystals. Relaxed defect configurations are obtained by taking a 12x12x12 supercell of a perfect periodic bulk crystal, inserting the point defect, and using force minimization to statically relax the atomic positions while keeping the system dimensions constant. Ef is computed by comparing the energy of the defect system to the same number of atoms in a perfect bulk crystal. pij is estimated as the difference in the system's global pressure with and without the defect multiplied by the system's volume.
Simple structural comparisons of the unrelaxed and relaxed defect configurations are used to help determine if the defect structure has relaxed to a different configuration. Relaxed structures that are identified as no longer consistent with the ideal defect definition are excluded from the table below. The only exception to this is if the lowest energy interstitial configuration does not coincide with a known ideal defect, its energies and pressure tensor are included under the listing "relaxed interstitial". The full list of calculation results including the transformed structures and the structural comparison values is included in the csv file available from the "Download raw data" link.
The calculation method used is available as the iprPy point_defect_static calculation method.
Notes and Disclaimers:
- These values are meant to be guidelines for comparing potentials, not the absolute values for any potential's properties. Values listed here may change if the calculation methods are updated due to improvements/corrections. Variations in the values may occur for changes in calculation methods, simulation software and implementations of the interatomic potentials.
- The computed formation energy and elastic dipole tensor values are sensitive to the size of the system used. The 12x12x12 supercell size was selected as it should provide a decent approximation of the true bulk values.
- The tests for identifying configuration relaxations are not guaranteed to be comprehensive or suitable for all point defect types. The table only shows the point defects that are consistent with the ideal configurations for that defect type, and the lowest energy interstitial.
- The "relaxed interstitial" label indicates that the structure is not consistent with any of the known ideal interstitial configurations. No information is provided as to what that relaxed strucutre is, whether it is an unknown structure, a "near"-ideal defect, or a collapse to amorphous.
- NIST disclaimer
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| 111 dumbbell | -66744.623 | -550.378 | -550.378 | -550.378 | -54.733 | -54.733 | -54.733 |
| tetrahedral interstitial | -55654.406 | -281.980 | -281.980 | -281.980 | -0.000 | -0.000 | -0.000 |
| crowdion interstitial | -16675.596 | -218.823 | -218.823 | -270.699 | 68.043 | 0.000 | 0.000 |
| 110 dumbbell | -11120.329 | -393.765 | -329.991 | -329.991 | 0.000 | 0.000 | 75.503 |
| vacancy | 0.482 | -1.904 | -1.904 | -1.904 | 0.000 | 0.000 | 0.000 |
| 2nn divacancy | 0.917 | -3.840 | -3.840 | -3.956 | 0.000 | 0.000 | 0.000 |
| 1nn divacancy | 0.929 | -4.765 | -3.537 | -3.537 | 0.000 | -0.000 | -0.239 |
| 100 dumbbell | 0.971 | 9.809 | 9.809 | 8.003 | 0.000 | -0.000 | -0.000 |
| octahedral interstitial | 1.024 | 9.791 | 9.791 | 9.791 | -0.000 | -0.000 | -0.000 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| relaxed interstitial | -16679.973 | -232.840 | -257.436 | -171.522 | -21.301 | 75.701 | -43.706 |
| 0001 dumbbell | -5557.580 | -212.276 | -212.276 | -198.228 | -0.000 | 0.000 | 0.000 |
| vacancy | 0.482 | -1.755 | -1.755 | -2.175 | -0.000 | 0.000 | -0.000 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| 111 dumbbell | -17026.451 | -27.930 | -27.930 | -27.930 | -7.155 | -7.155 | -7.155 |
| tetrahedral interstitial | -14162.443 | -52.744 | -52.744 | -52.744 | -0.000 | -0.000 | -0.000 |
| 110 dumbbell | -2827.304 | 14.594 | -9.853 | -9.853 | 0.000 | 0.000 | -5.338 |
| vacancy | 2.489 | 0.101 | 0.101 | 0.101 | -0.000 | -0.000 | 0.000 |
| octahedral interstitial | 3.522 | -2.278 | -2.278 | -2.278 | -0.000 | 0.000 | 0.000 |
| 1nn divacancy | 4.570 | 0.175 | 0.184 | 0.184 | -0.000 | -0.000 | -0.002 |
| 2nn divacancy | 4.985 | 0.205 | 0.205 | 0.194 | -0.000 | -0.000 | 0.000 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| tetrahedral interstitial | -14158.472 | -77.516 | -95.880 | -77.516 | 0.000 | -0.000 | 0.000 |
| crowdion interstitial | -4214.020 | -46.420 | -46.420 | -46.420 | -0.767 | -0.767 | -0.767 |
| octahedral interstitial | -4206.851 | -32.783 | -19.416 | -19.416 | 0.000 | 0.000 | -0.000 |
| 111 dumbbell | -2799.536 | -59.442 | -59.442 | -59.442 | 8.147 | 8.147 | 8.147 |
| 110 dumbbell | 1.775 | 29.286 | 29.630 | 29.630 | -0.000 | -0.000 | -0.496 |
| 100 dumbbell | 2.023 | 32.071 | 32.071 | 30.779 | 0.000 | 0.000 | 0.000 |
| vacancy | 2.725 | -17.705 | -17.705 | -17.705 | -0.000 | 0.000 | -0.000 |
| 1nn divacancy | 5.364 | -35.392 | -35.392 | -35.392 | 0.054 | 0.054 | 0.054 |
| 2nn divacancy | 5.716 | -35.213 | -35.213 | -35.509 | 0.000 | 0.000 | -0.000 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| 110 dumbbell | -8492.591 | -0.094 | 1.266 | 1.266 | 0.000 | 0.000 | -15.009 |
| crowdion interstitial | -4252.135 | 5.868 | 5.868 | 5.868 | -4.229 | -4.229 | -4.229 |
| octahedral interstitial | -4250.290 | 13.746 | 19.627 | 19.627 | -0.000 | 0.000 | -0.000 |
| 111 dumbbell | -2829.810 | 5.214 | 5.214 | 5.214 | -7.114 | -7.114 | -7.114 |
| vacancy | 1.703 | 0.369 | 0.369 | 0.369 | 0.000 | -0.000 | -0.000 |
| 2nn divacancy | 3.111 | 3.334 | 3.334 | -5.234 | 0.000 | 0.000 | 0.000 |
| 1nn divacancy | 3.449 | 0.958 | 0.958 | 0.958 | 1.324 | 1.324 | 1.324 |
| 100 dumbbell | 6.434 | 1.024 | 1.024 | -4.356 | -0.000 | 0.000 | -0.000 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| bond centered interstitial | -4253.148 | -2.862 | -2.862 | -2.862 | 2.374 | 2.374 | 2.374 |
| vacancy | 0.876 | -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -0.000 |
| 1nn divacancy | 1.314 | -0.000 | -0.000 | -0.000 | -0.000 | -0.000 | -0.000 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| vacancy | 1.890 | -7.798 | -7.798 | -7.798 | 0.000 | 0.000 | 0.000 |
| 2nn divacancy | 3.749 | -15.507 | -15.507 | -15.340 | 0.000 | 0.000 | 0.000 |
| 1nn divacancy | 3.782 | -15.917 | -15.276 | -15.276 | 0.000 | 0.000 | 0.885 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| tetrahedral interstitial | -1.744 | -8.964 | -8.964 | -8.964 | 0.000 | 0.000 | 0.000 |
| 111 dumbbell | -1.500 | -1.928 | -1.928 | -1.928 | 3.153 | 3.153 | 3.153 |
| 110 dumbbell | 0.617 | 4.950 | -1.953 | -1.953 | -0.000 | -0.000 | 4.874 |
| vacancy | 1.914 | -0.079 | -0.079 | -0.079 | -0.000 | -0.000 | -0.000 |
| 1nn divacancy | 3.556 | 0.006 | -0.258 | -0.258 | 0.000 | 0.000 | -0.010 |
| 2nn divacancy | 3.788 | -0.148 | -0.148 | 0.182 | -0.000 | 0.000 | -0.000 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| tetrahedral interstitial | -2.801 | -2.701 | 4.879 | -2.701 | 0.000 | 0.000 | 0.000 |
| 110 dumbbell | -2.736 | -6.944 | 0.277 | 0.277 | 0.000 | 0.000 | -10.544 |
| vacancy | 1.755 | 0.518 | 0.518 | 0.518 | -0.000 | 0.000 | -0.000 |
| 2nn divacancy | 3.170 | 1.802 | 1.802 | -0.053 | -0.000 | 0.000 | -0.000 |
| 1nn divacancy | 3.437 | 0.649 | 0.649 | 0.649 | 0.303 | 0.303 | 0.303 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| vacancy | 1.352 | -8.816 | -8.816 | -10.963 | 0.000 | 0.000 | -0.000 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| vacancy | 1.914 | -0.082 | -0.082 | -0.079 | 0.000 | -0.000 | -0.000 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| tetragonal interstitial | -1.599 | -10.706 | -10.706 | -10.706 | -0.000 | -0.000 | -0.000 |
| vacancy | 1.618 | -10.120 | -10.120 | -10.120 | -0.000 | -0.000 | -0.000 |
| 1nn divacancy | 3.255 | -17.996 | -17.996 | -17.996 | -0.524 | -0.524 | -0.524 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| relaxed interstitial | -8351.262 | -11962.682 | -12229.773 | -12095.970 | 198.125 | 210.447 | -160.863 |
| tetragonal interstitial | -2.347 | 1.293 | 1.293 | 1.293 | 0.000 | 0.000 | 0.000 |
| vacancy | 0.790 | -0.037 | -0.037 | -0.037 | 0.000 | 0.000 | 0.000 |
| 1nn divacancy | 1.409 | -1.128 | -1.128 | -1.128 | -3.653 | -3.653 | -3.653 |
| Point Defect | Ef (eV) | p11 (eV) | p22 (eV) | p33 (eV) | p12 (eV) | p13 (eV) | p23 (eV) |
|---|---|---|---|---|---|---|---|
| vacancy | 0.832 | -0.017 | -0.017 | -0.017 | 0.000 | 0.000 | 0.000 |
| 1nn divacancy | 1.248 | -0.024 | -0.024 | -0.024 | -0.000 | -0.000 | -0.000 |
Dislocation Monopole Structures
Plots of the atomic structure of dislocation cores are shown here. The dislocations were constructed using dislocation monopole configurations in which a straight dislocation is created in the center of the system using anisotropic elasticity and the elastic constants computed above. A cylindrical region centered around the dislocation is allowed to relax while the remaining atoms are held fixed. The relaxation consists of 1 million NVT steps at 10 K followed by an energy/force minimization. The final plots show the atomic configuration near the center of the monopole system, and display differential displacement arrows and Nye tensor color maps that are both taken with respect to the Burgers vector direction.
These plots show a wide range of final structures, some corresponding to expected dislocation structures and others not. While the characterizations are dislocation-type dependent, some general classification guidelines are listed here.
- Planar dislocations exist on a single slip plane, which results in a grouping of large differential displacement arrows between two atomic rows. All edge dislocations and some screw dislocations may be planar dislocations.
- Non-planar dislocations are screw dislocations that are positioned at the intersection of more than one slip plane. This results in the differential displacement arrows circling the position of the dislocation core. These core structures often have rotational symmetry due to the intersecting planes.
- The plotted atomic positions are also useful for characterizing the final configuration. For screw dislocations, the atomic projections should match projections in the bulk crystal if the crystal has not transformed. For edge dislocations, the projections should be the bulk projections with the extra half-plane of atoms directly above the dislocation core.
- Images in which the atomic positions look correct but no large differential displacement arrows are observed are likely the result of the dislocation gliding out of view during relaxation.
- Phase transformations are identified by large differential displacement arrows spread out in both plot directions, often across the whole image. Additionally, the atomic position projections will likely not correspond to what is expected.
- Twins typically appear as a long line of large differential displacement arrows along a single plane. Note that twins can nucleate phase transformations so the distinction between the two is not always clear.
The calculation method used is available as the iprPy dislocation_monopole calculation method.
Links are provided above the images allowing for users to download the final defect configuration and a corresponding defect-free configuration if they wish to further analyze the structures or use them for subsequent simulations.
Notes and Disclaimers:
- While the general dislocation monopole method is universal to all dislocations, these calculations rely on dislocation-type specific atomic configuration sizes and differential displacement/Nye tensor settings. For the system size, the specific super cell multipliers vary based on the crystal and crystal vector orientations, but the sizes are always selected to minimize the length along the dislocation line and attempt to make the two other box dimensions similar. For the structure analysis tools, both are sensitive to the cutoff distance used to identify neighbors, and both have some method-specific parameters. Where known, the settings used here correspond to the most common ones used in literature.
- For bcc dislocations, the differential displacement vectors shown here are "wrapped" in that if their magnitudes exceed b/2 then a value of b is added/subtracted to wrap the vector back around. This convention reveals the dislocation's structure and location without showing the slip history.
- NIST disclaimer
Version Information:
- 2025-08-07. Plots first added.

MD Solid Property Predictions
Plots of lattice and elastic constants are shown as a function of temperature. The 0K points were taken from the Crystal Structure Predictions and the Elastic Constants Predictions sections above for the unique crystal structures relaxed with the "dynamic" method. Starting from the 0 K relaxed crystal unit cells, supercell systems are created by replicating all three dimensions by the same multiplier to achieve at least 4000 atoms. The systems are then relaxed at 50 K and zero pressure using 1 million NPT steps. Lattice constants are estimated by averaging the measured box dimensions. Temperatures are iteratively increased by 50 K, with each subsequent relaxation calculation starting from the final atomic configuration at the previous temperature and relaxing for another 1 million steps.
The elastic constants are calculated using the deformation–fluctuation hybrid method. Starting from the final atomic configurations of the dynamic relaxations, the system is allowed to evolve at constant volume with a Langevin thermostat. The Born matrix is computed during this run by evaluating how the atomic forces would vary due to applied linear strain fields. The elastic constants can then be estimated using the averaged Born matrix values and the averaged stresses on the system.
The calculation methods used are available as the iprPy relax_dynamic and elastic_constants_dynamic calculation methods.
Clicking on the image of a plot will open an interactive version of it in a new tab. The underlying data for the plots can be downloaded by clicking on the links above each plot.
Notes and Disclaimers:
- The maximum temperature shown for a crystal either corresponds to the maximum value that has been computed so far, or the maximum value that the structure is believed to have remained untransformed. The unstable/transformation temperature identification is mostly automated and can miss transitions not associated with large discontinuities in property values with changing temperature.
- For succinctness, the elastic constants displayed here are averaged according to the 0 K structure's crystal symmetry family. If the structures deviate from the expected symmetry at higher temperatures, the values may not be valid. Raw results are available for verification if requested.
- NIST disclaimer
Version Information:
- 2025-08-07. Plots added.
Click on plot to load interactive version
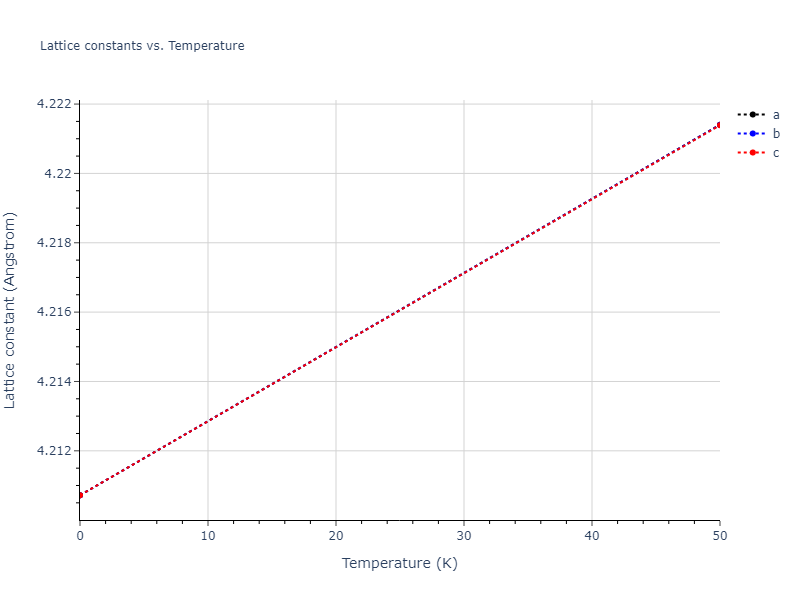
Click on plot to load interactive version
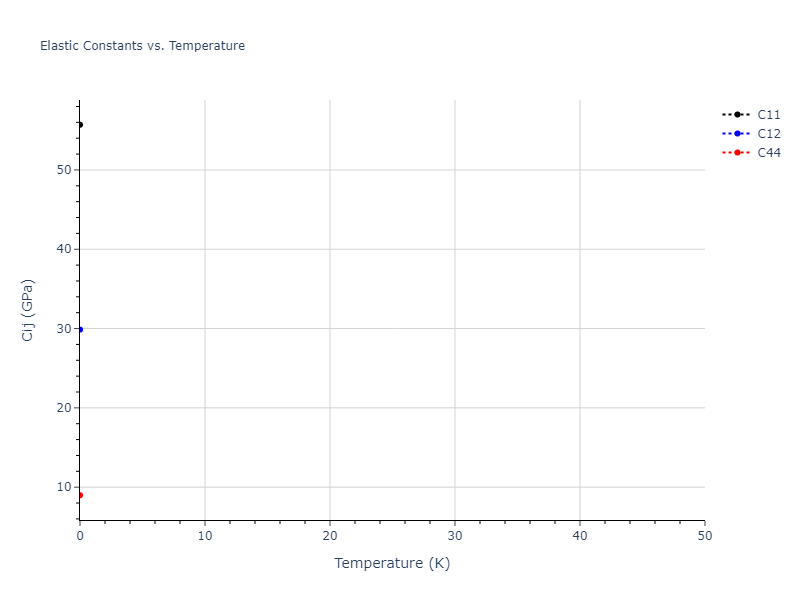
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
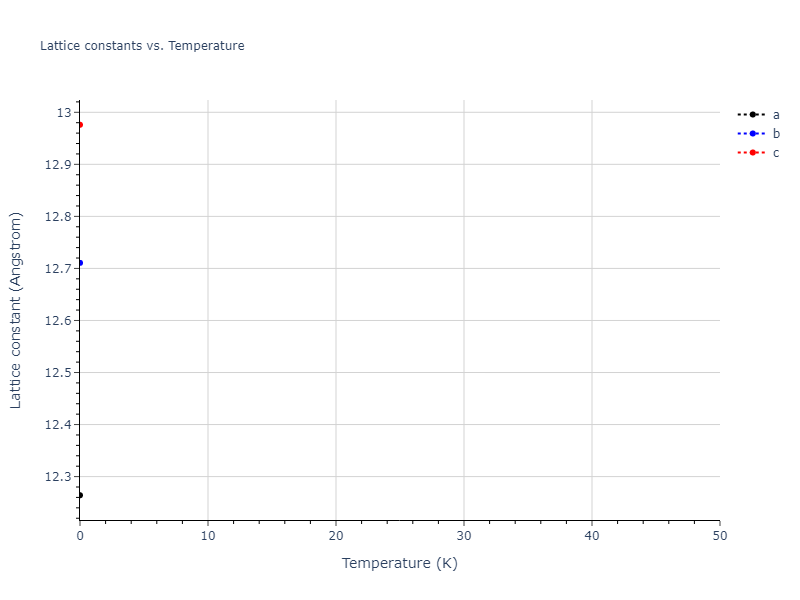
Click on plot to load interactive version
Click on plot to load interactive version
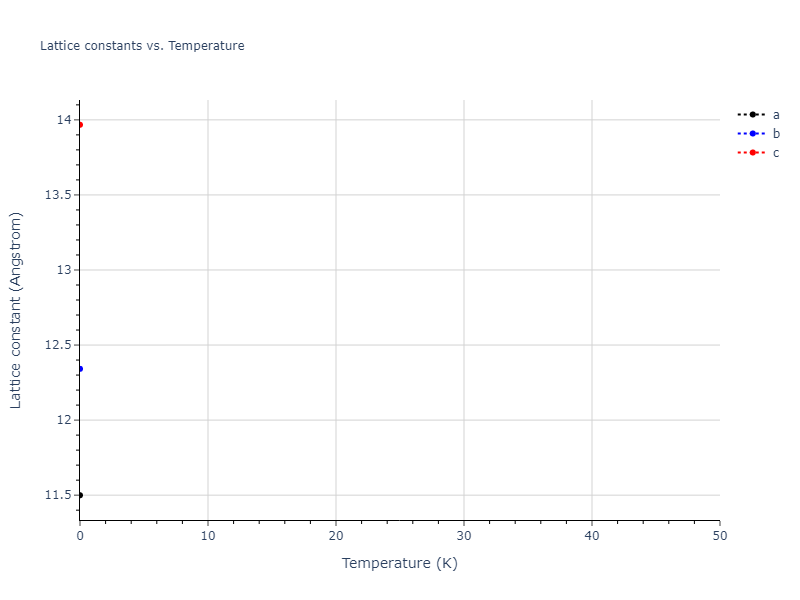
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
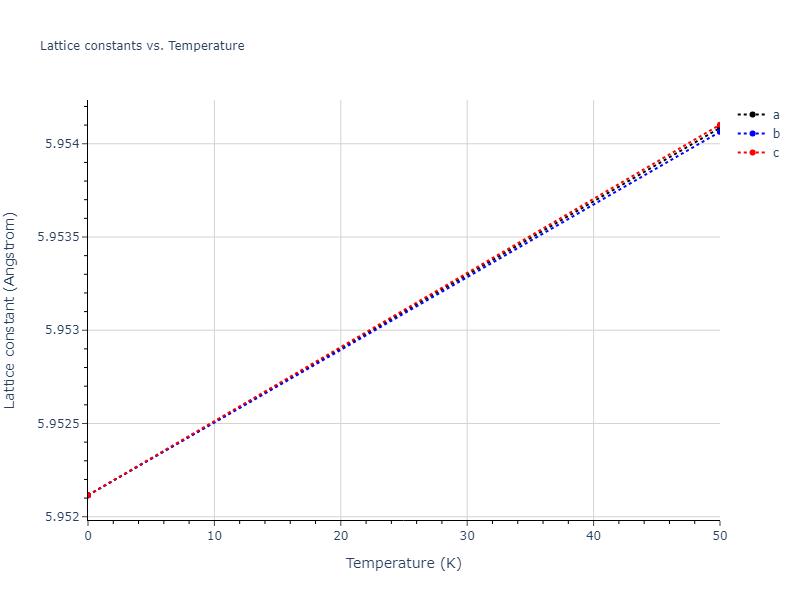
Click on plot to load interactive version
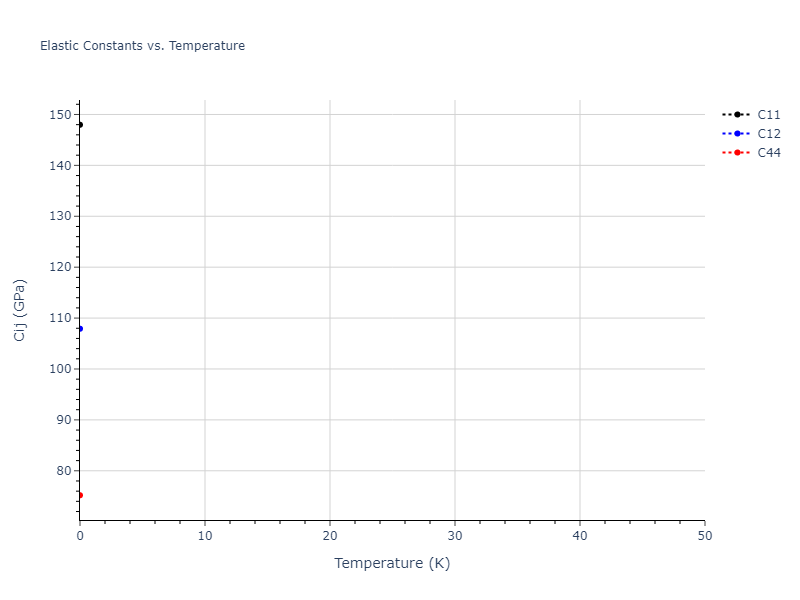
Click on plot to load interactive version
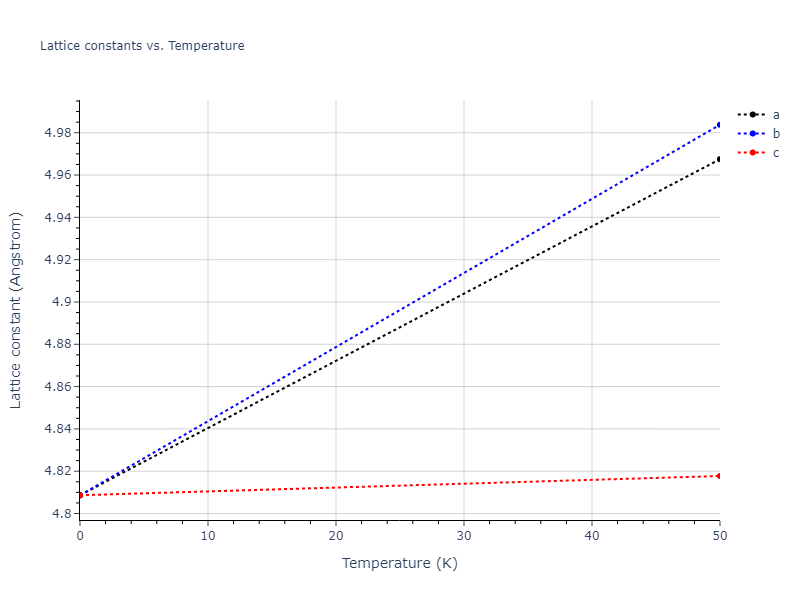
Click on plot to load interactive version
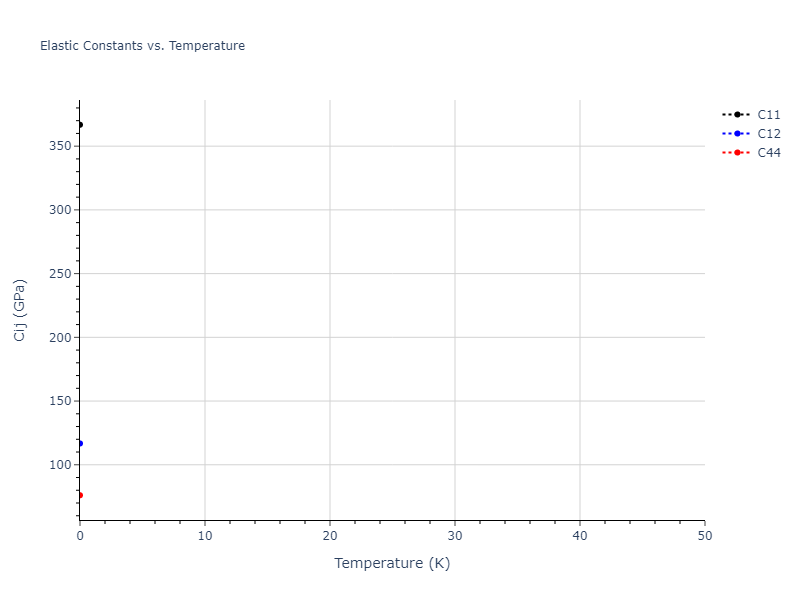
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
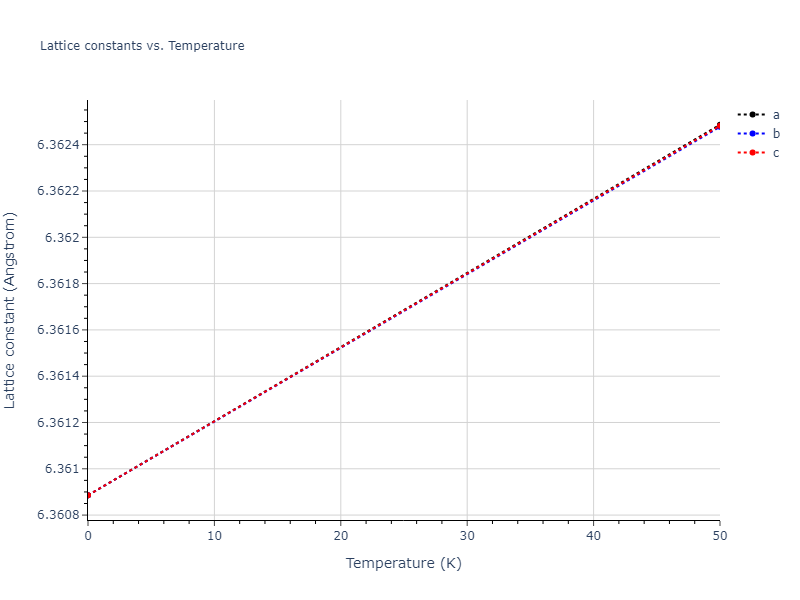
Click on plot to load interactive version
Click on plot to load interactive version
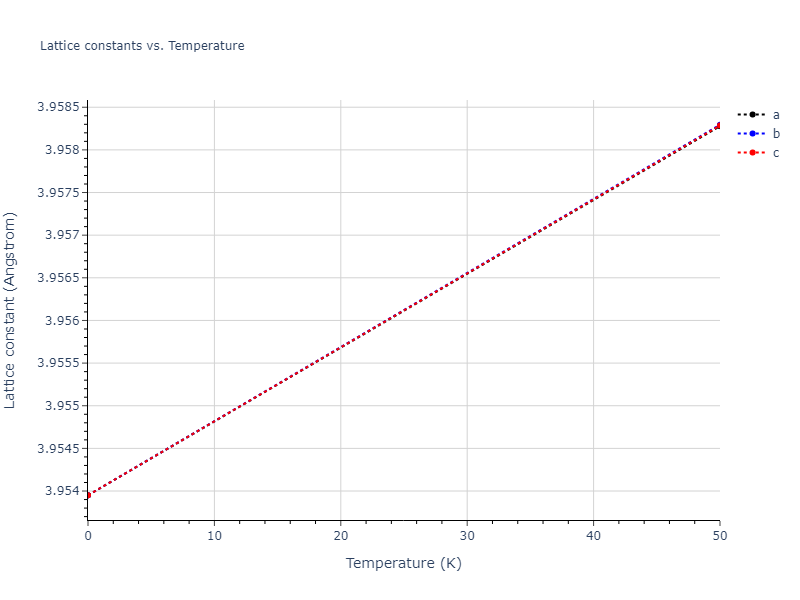
Click on plot to load interactive version
Click on plot to load interactive version
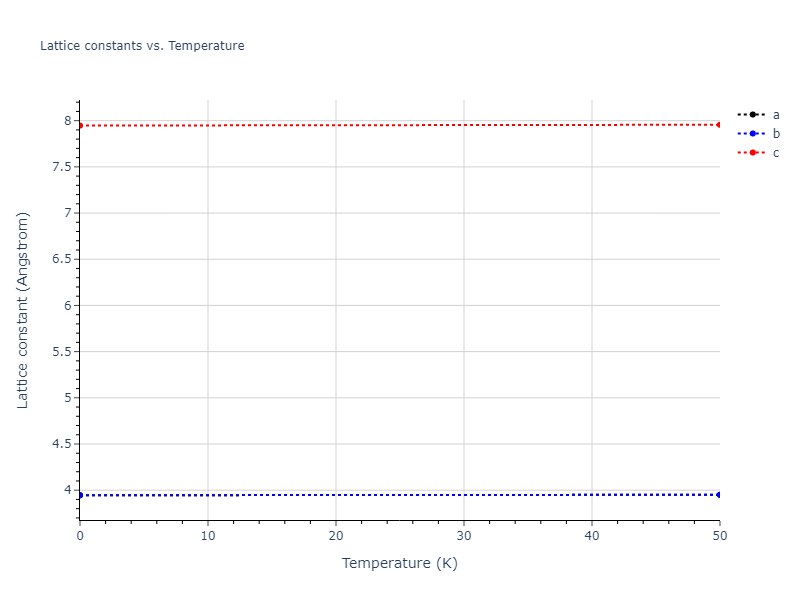
Click on plot to load interactive version
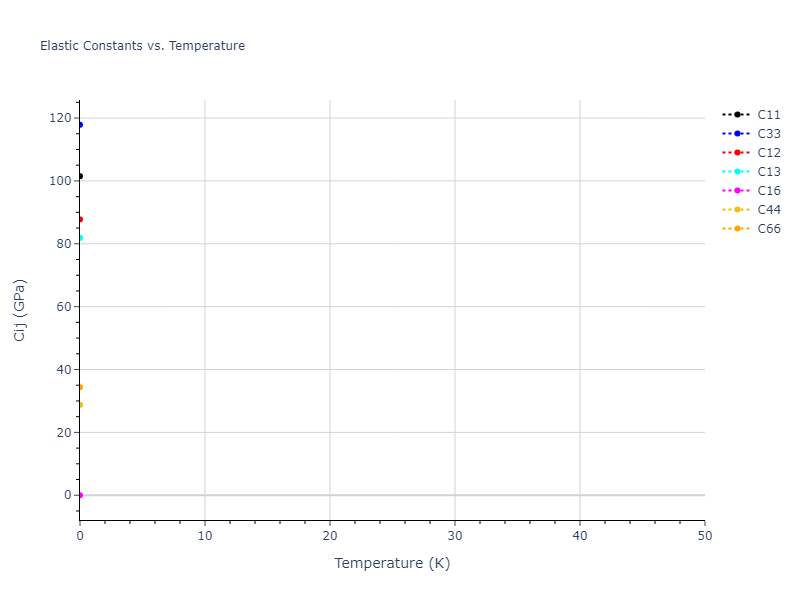
Click on plot to load interactive version
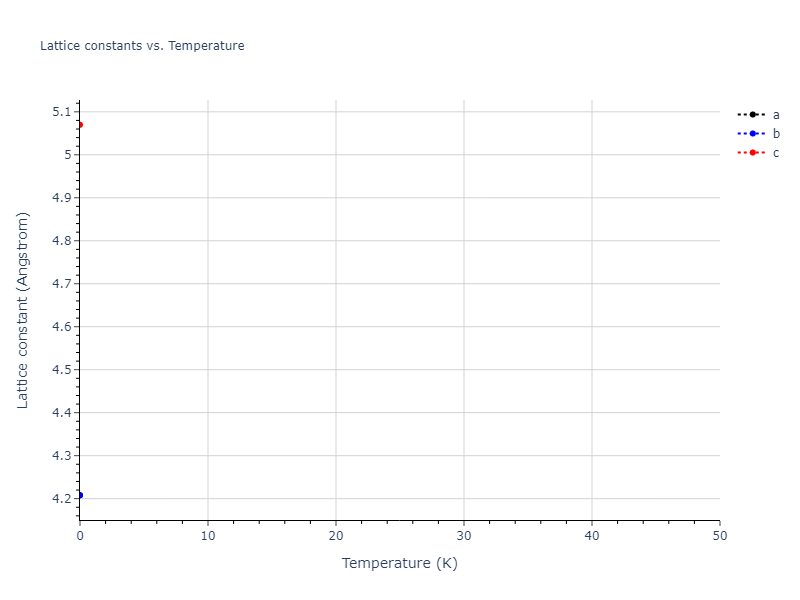
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
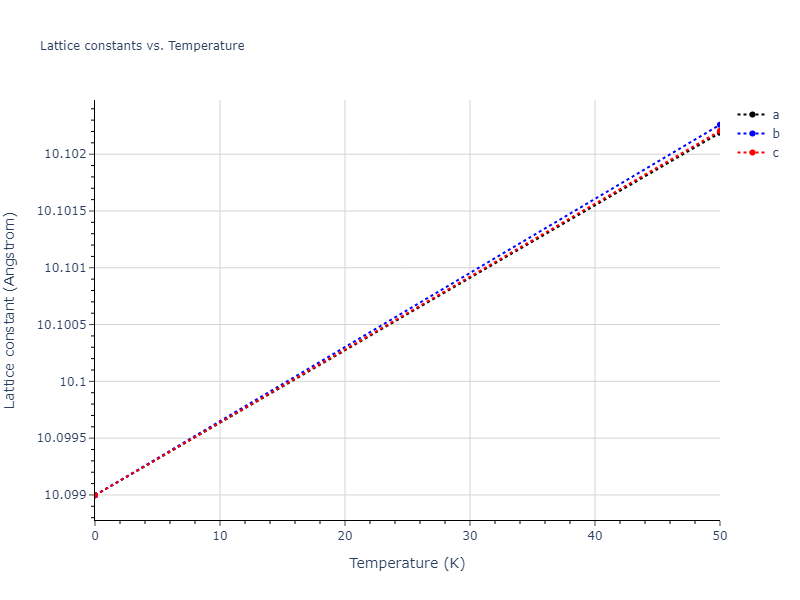
Click on plot to load interactive version
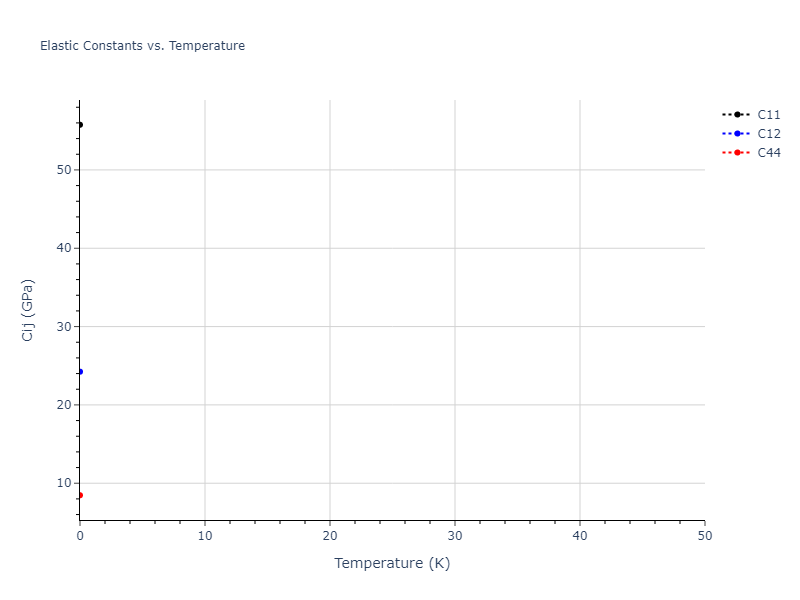
Click on plot to load interactive version
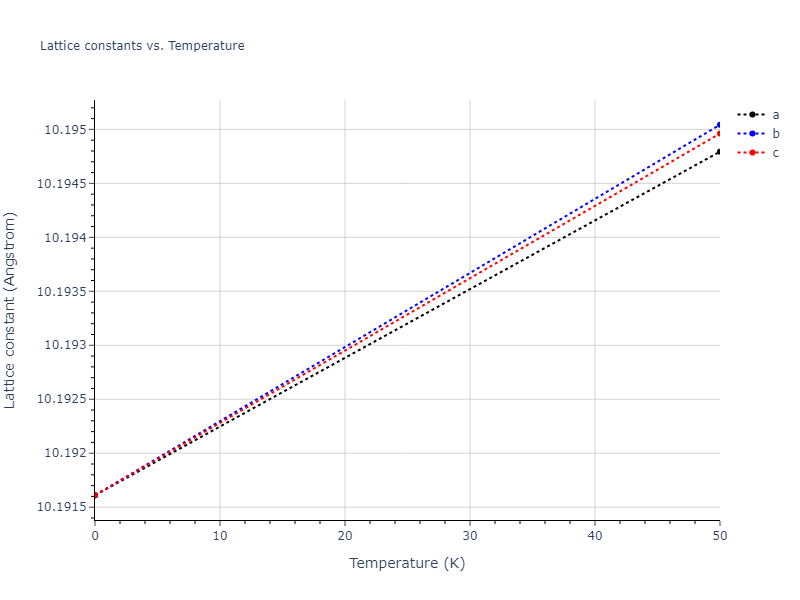
Click on plot to load interactive version
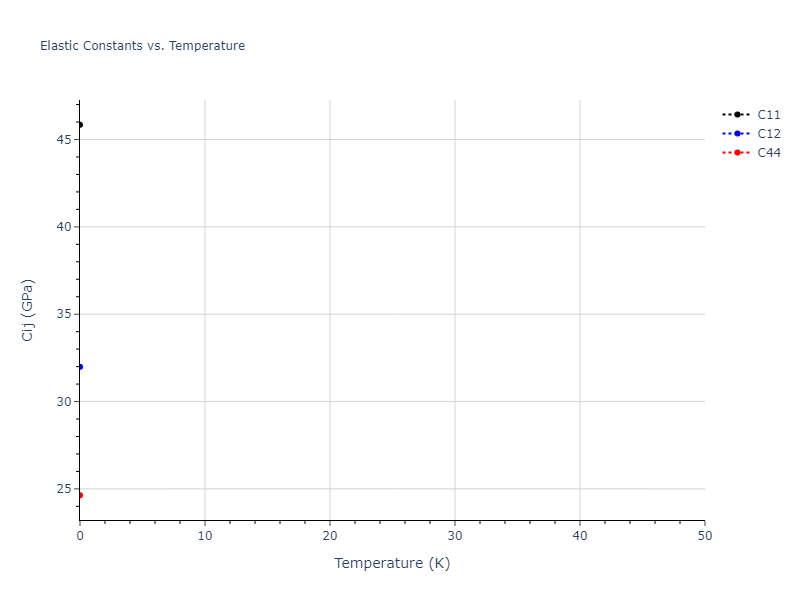
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
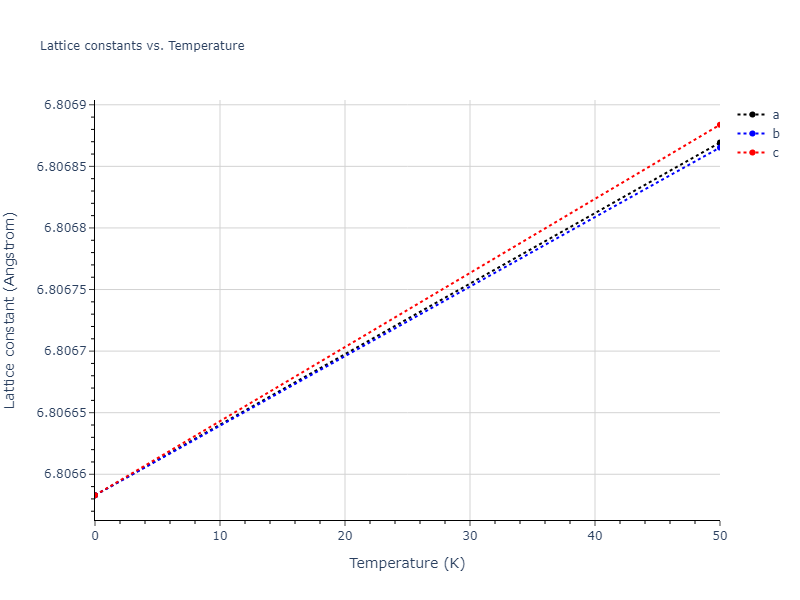
Click on plot to load interactive version
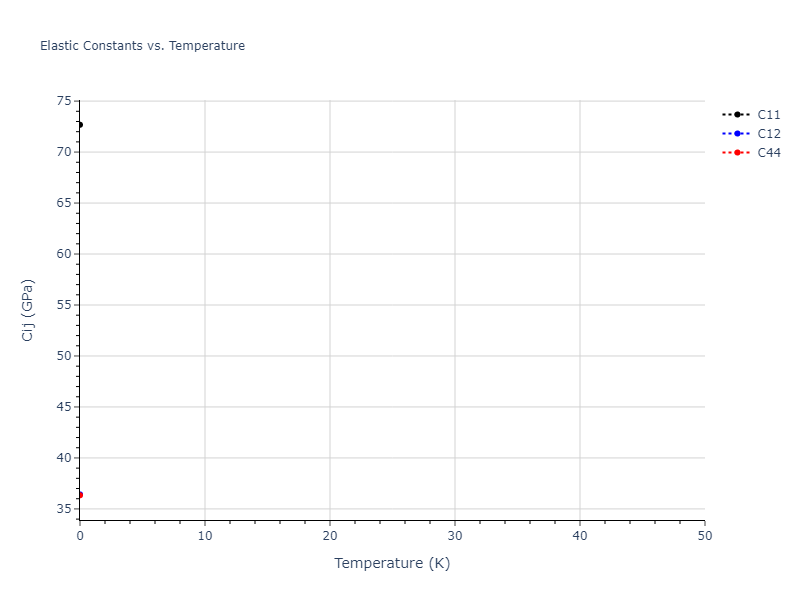
Click on plot to load interactive version
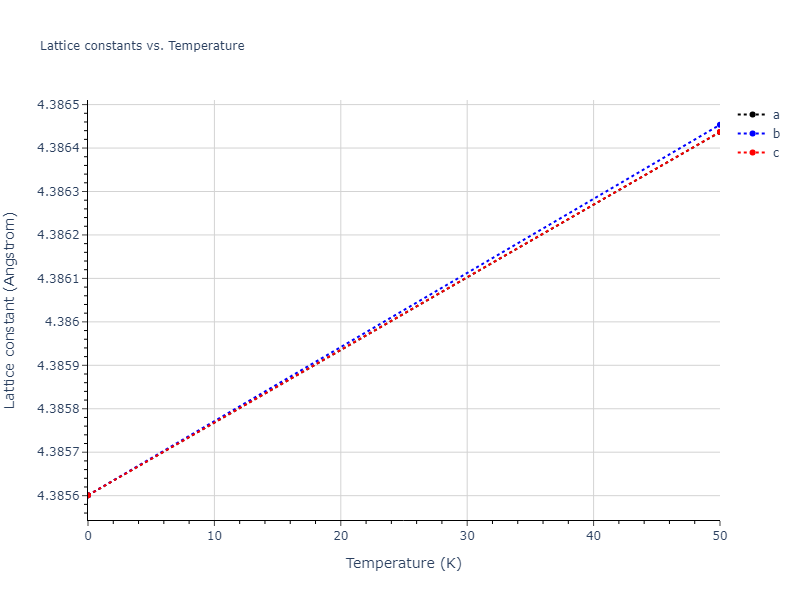
Click on plot to load interactive version
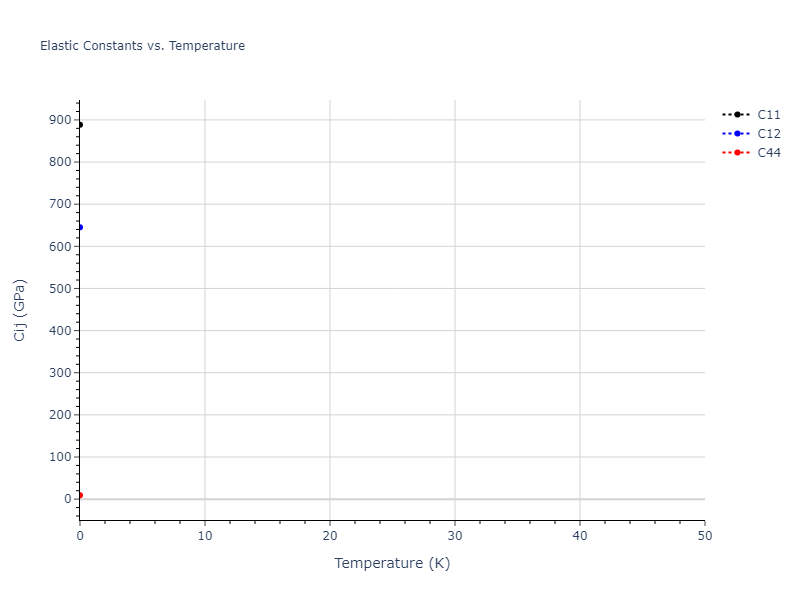
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
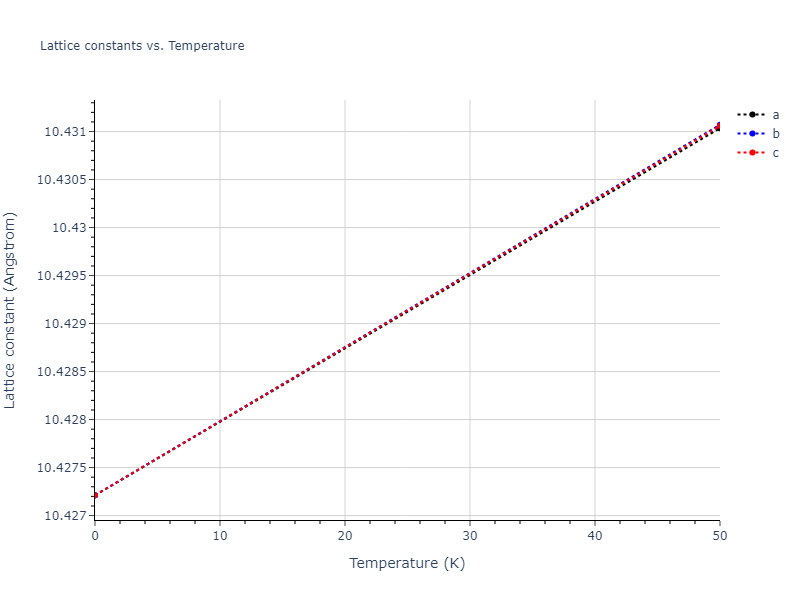
Click on plot to load interactive version
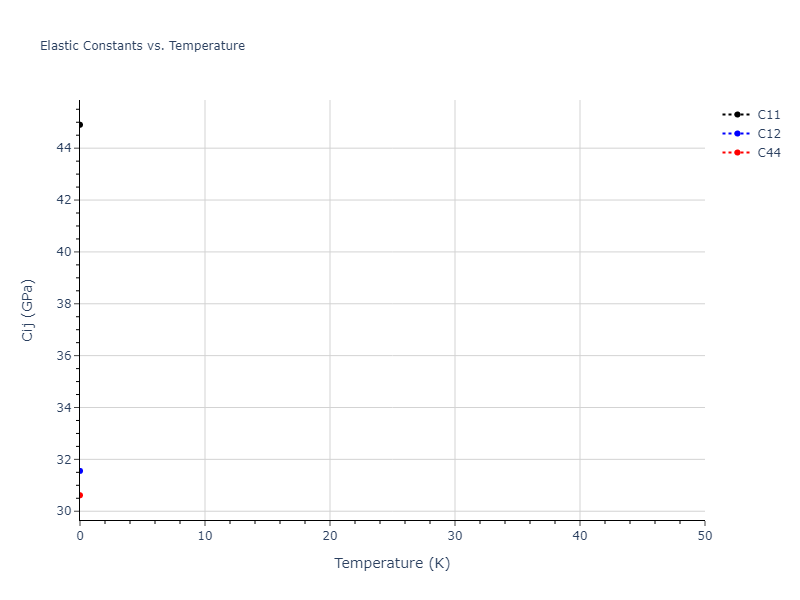
Click on plot to load interactive version
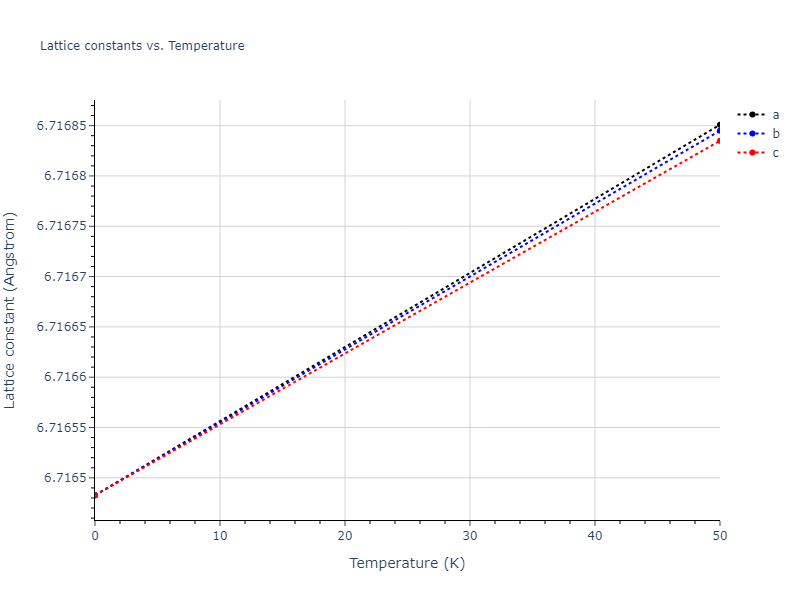
Click on plot to load interactive version
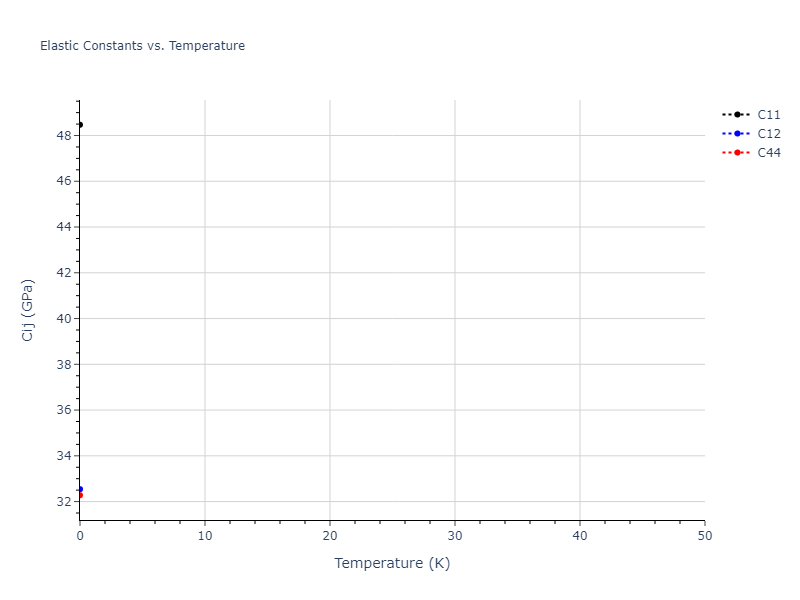
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
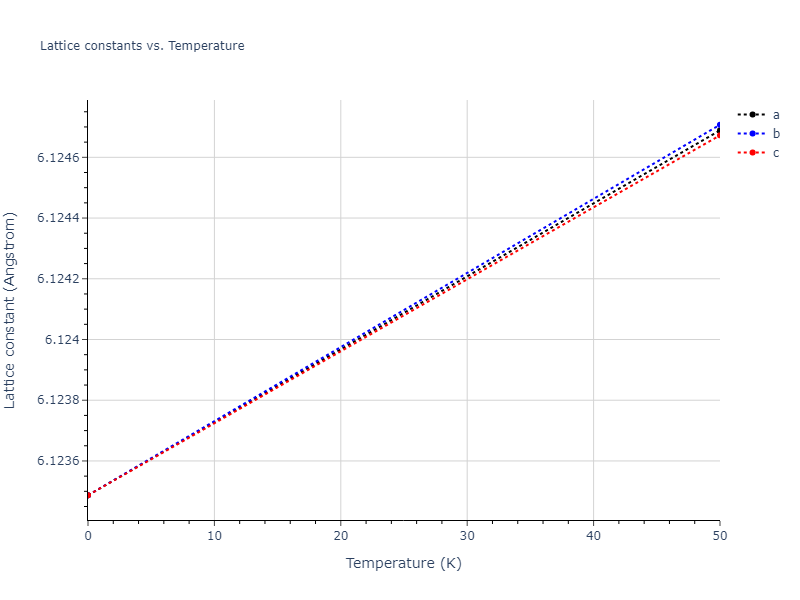
Click on plot to load interactive version
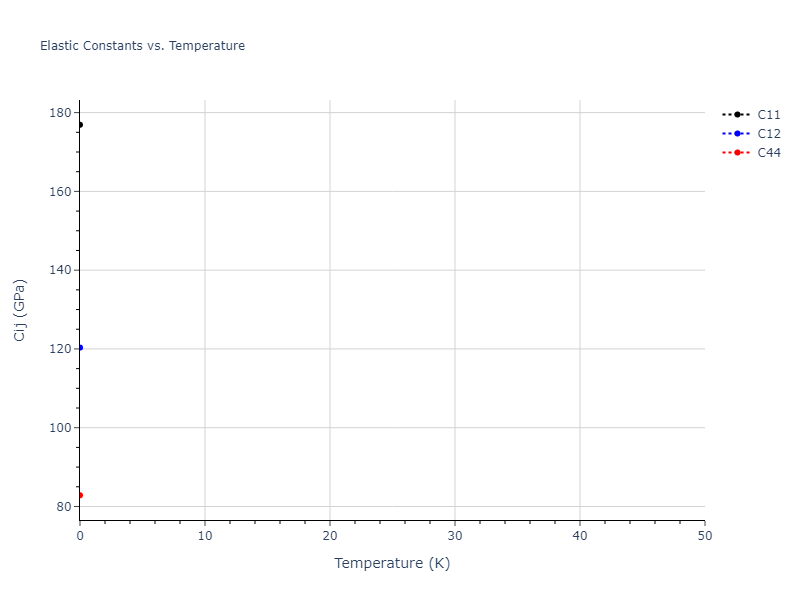
Click on plot to load interactive version
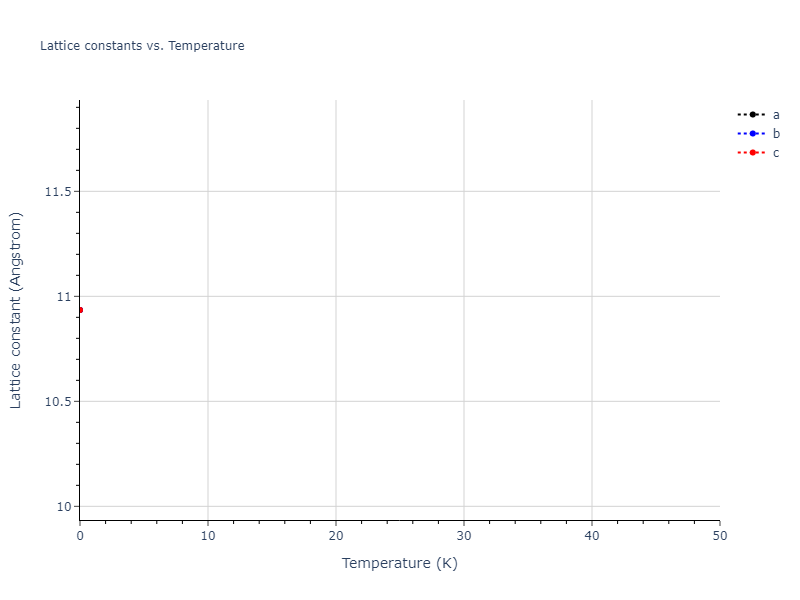
Click on plot to load interactive version
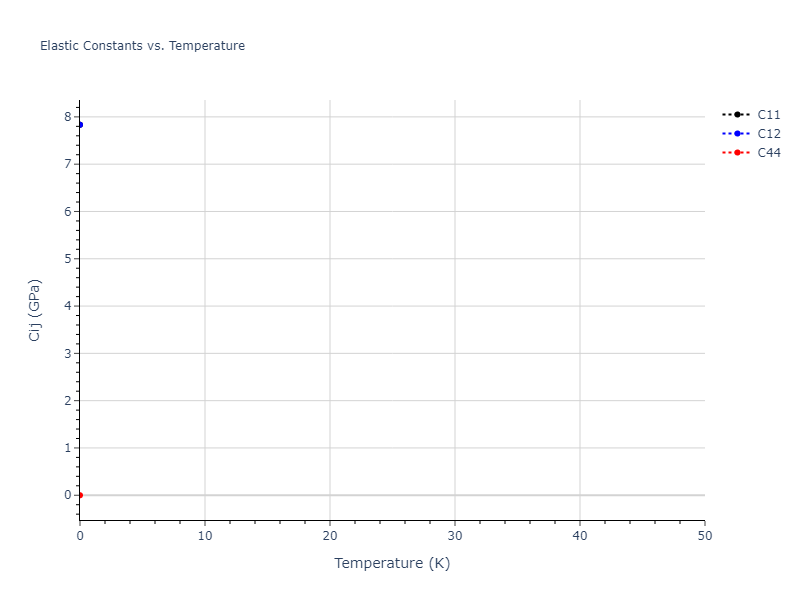
Click on plot to load interactive version
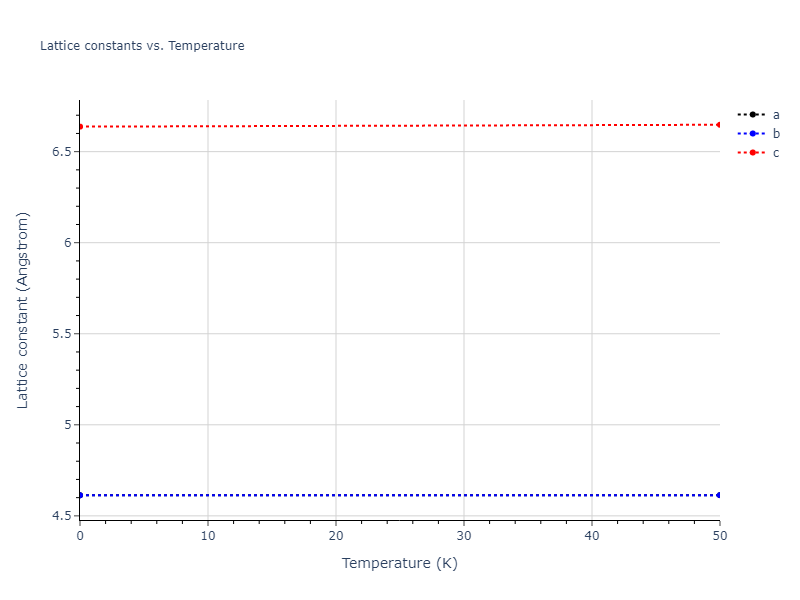
Click on plot to load interactive version
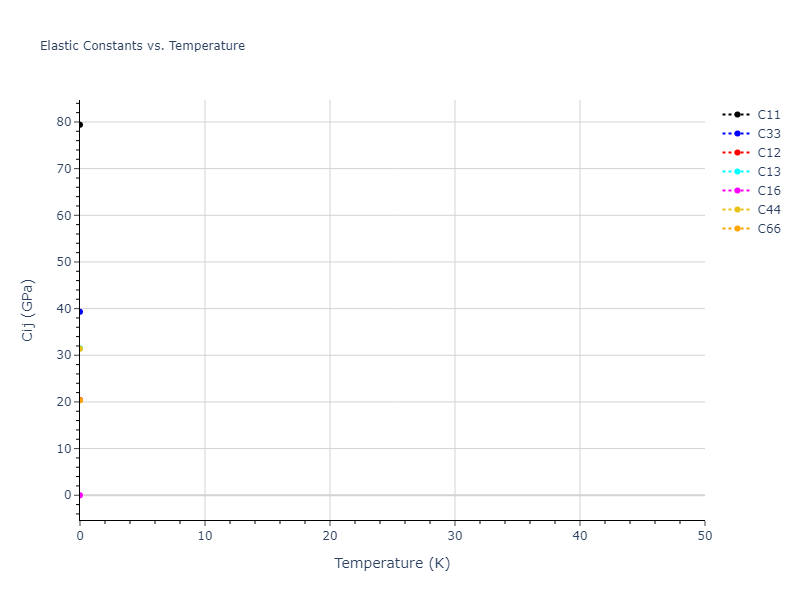
Click on plot to load interactive version
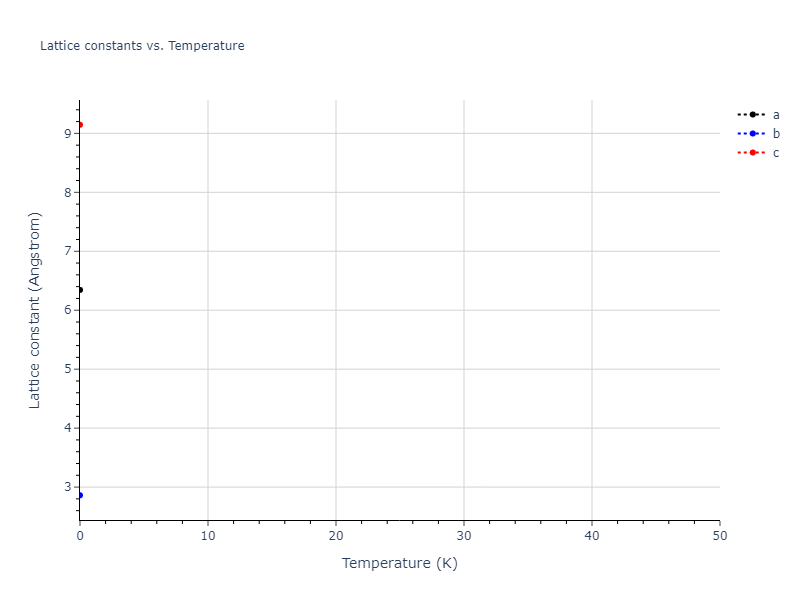
Click on plot to load interactive version
Click on plot to load interactive version
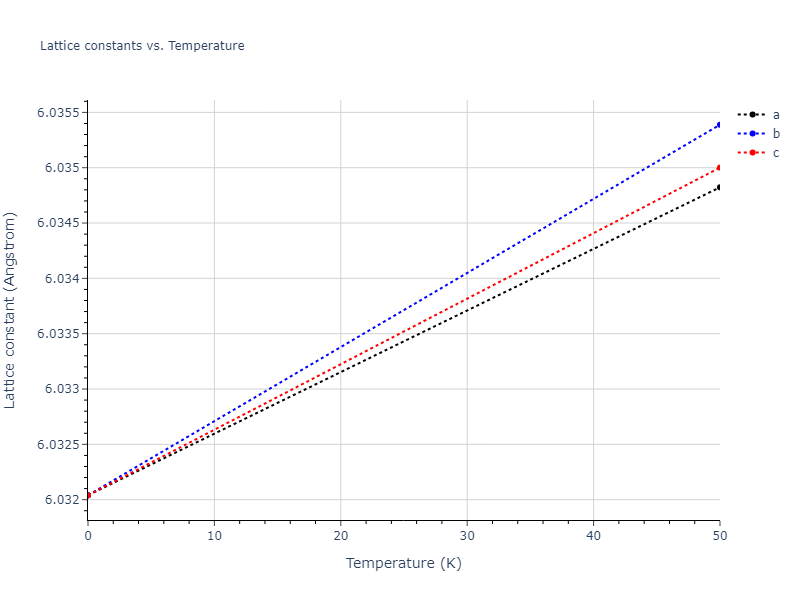
Click on plot to load interactive version
Click on plot to load interactive version
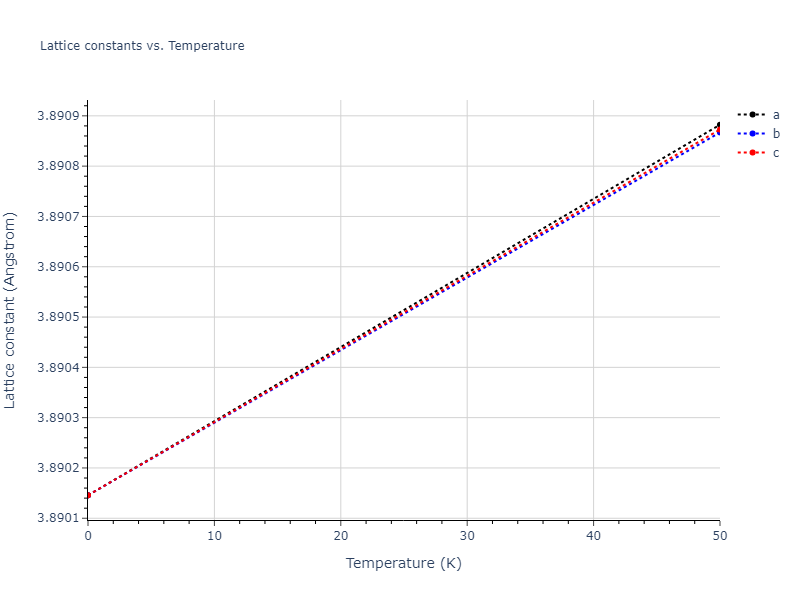
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
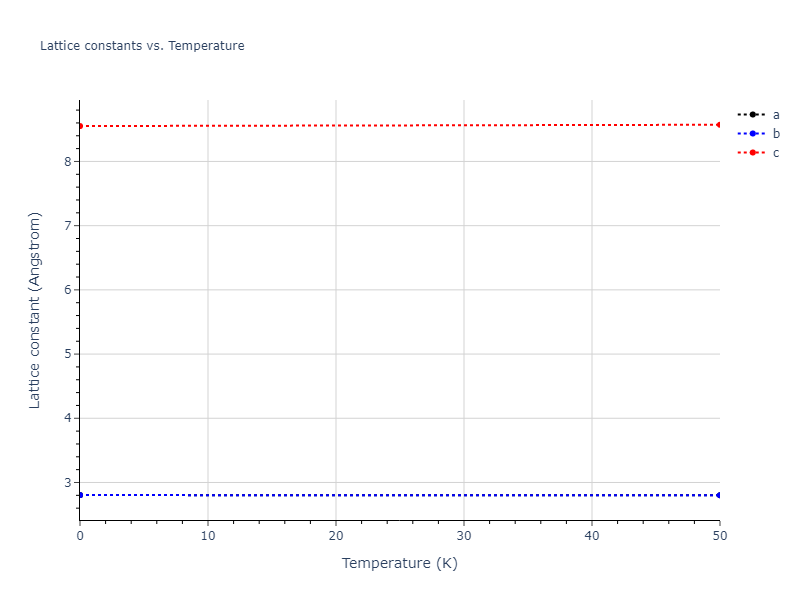
Click on plot to load interactive version
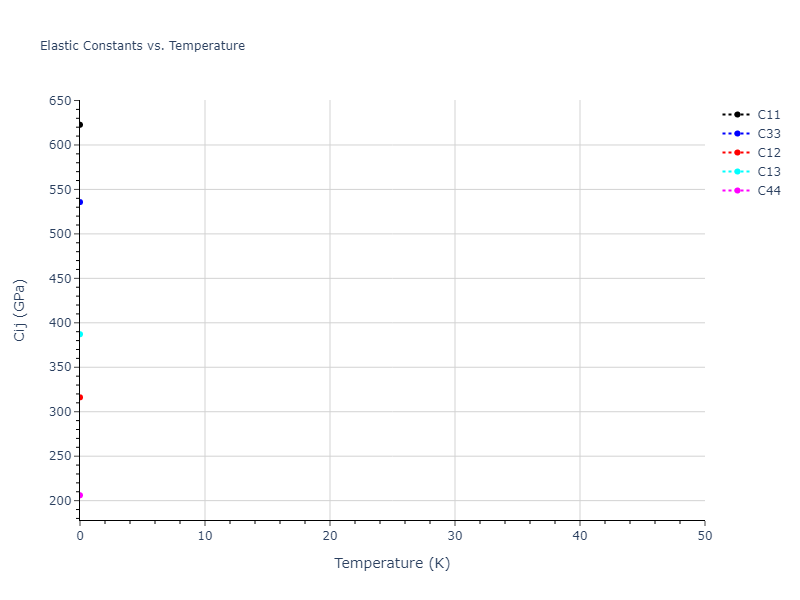
Click on plot to load interactive version
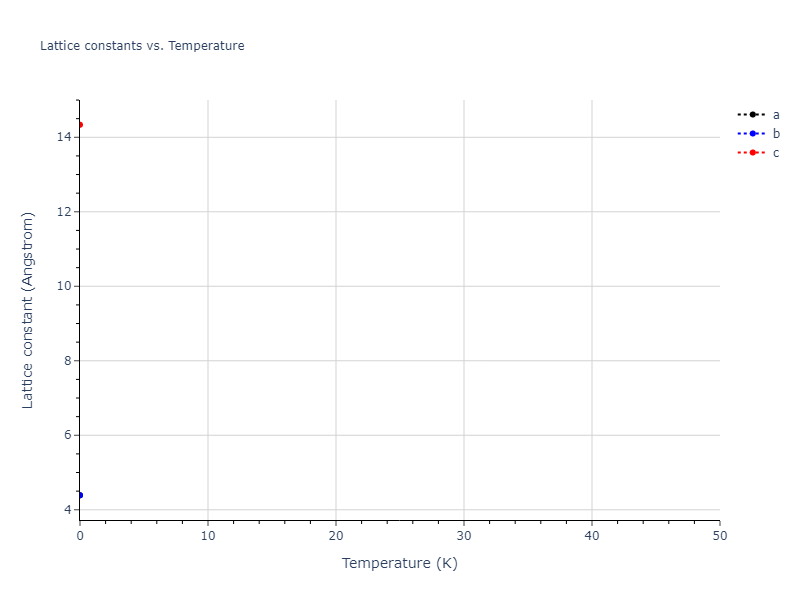
Click on plot to load interactive version
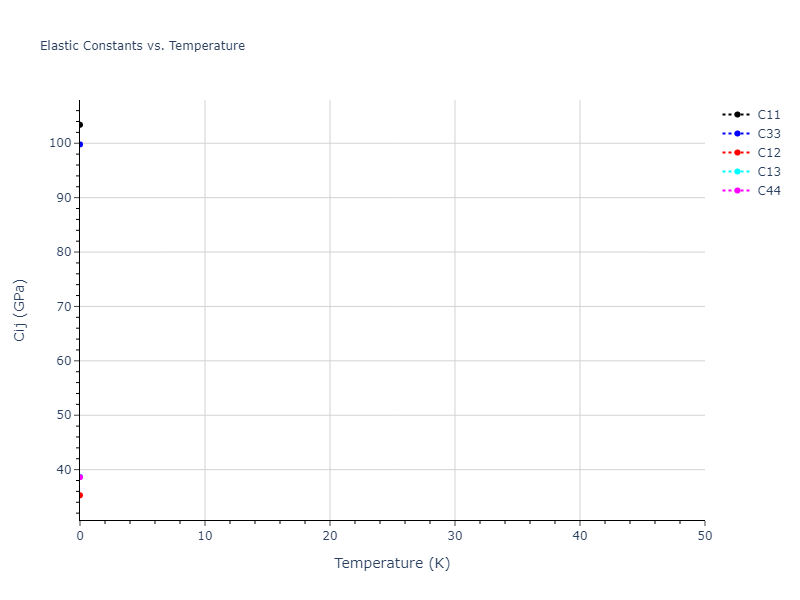
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
Click on plot to load interactive version
MD Thermodynamic Predictions
Plots of internal energy, Gibbs free energy, entropy, heat capacity and volume are shown here as a function of temperature for various crystal structures and liquid phases. The included crystal structures correspond to those in the Solid Structures vs. Temperature section, and the liquid phases to those in the Liquid Properties section. Internal energy and volume are taken from the associated structure relaxations mentioned in those previous sections. Constant pressure heat capacity is estimated using 3-point numerical derivatives of enthalpy versus temperature. Note that since all simulations done here are at 0 pressure, internal energy and enthalpy are equivalent.
The Gibbs energies of the phases are estimated using thermodynamic integration between the interatomic potential in question and a simpler model with known Gibbs free energy values. For solids, the reference model is an Einstein solid, while for liquids it is the Uhlenbeck-Ford potential. Besides a short run at the start of the solid calculations to estimate Einstein model spring constants, the two calculations proceed similarly. Starting with the final relaxation configurations, the systems are stabilized for 25,000 steps. Then, over the next 50,000 steps the potential is swapped out for the reference potential. The system is then stabilized for another 25,000 steps with the reference model before a reverse swap of 50,000 steps is performed. The simulation ends with one final 25,000 step stabilization period. From the simulation, the irreversible work of transformation is estimated and used to compute the absolute Gibbs free energy of the target phase and potential relative to the reference potential. Entropy is estimated as the difference in enthalpy and Gibbs free energy and divided by temperature.
The calculation methods used are available as the iprPy relax_dynamic, relax_liquid_redo, free_energy, and free_energy_liquid calculation methods.
Clicking on the image of a plot will open an interactive version of it in a new tab. The underlying data for the plots can be downloaded by clicking on the links above each plot.
Notes and Disclaimers:
- Good free energy estimates require that the initial and final phases before and after the thermodynamic integration remain the same. This is not an issue with the liquid calculations as the Uhlenbeck-Ford model always predicts a liquid, but may be an issue for the solid phase estimates. This is likely to be an issue for non-compact phases which the Einstein model is poor at representing.
- NIST disclaimer
Version Information:
- 2025-08-07. Plots added.