bond_angle_scan - Methodology and code
Python imports
[1]:
# Standard library imports
from pathlib import Path
import datetime
from math import floor
from typing import Optional
# http://www.numpy.org/
import numpy as np
# https://ipython.org/
from IPython.display import display, Markdown
# https://github.com/usnistgov/atomman
import atomman as am
import atomman.lammps as lmp
import atomman.unitconvert as uc
from atomman.tools import filltemplate, aslist
# https://github.com/usnistgov/iprPy
import iprPy
from iprPy.tools import read_calc_file
print('Notebook last executed on', datetime.date.today(), 'using iprPy version', iprPy.__version__)
Notebook last executed on 2023-07-31 using iprPy version 0.11.6
1. Load calculation and view description
1.1. Load the calculation
[2]:
# Load the calculation being demoed
calculation = iprPy.load_calculation('bond_angle_scan')
1.2. Display calculation description and theory
[3]:
# Display main docs and theory
display(Markdown(calculation.maindoc))
display(Markdown(calculation.theorydoc))
bond_angle_scan calculation style
Lucas M. Hale, lucas.hale@nist.gov, Materials Science and Engineering Division, NIST.
Introduction
The bond_angle_scan calculation style evaluates the interaction energy between three atoms at varying distances and angles. This provides a means of characterizing the three-body interactions of a given potential. These interactions can provide insight into the bonding predictions for a potential as well as a means of fingerprinting the potentials.
Version notes
2021-04-XX: Calculation added
Additional dependencies
Disclaimers
Method and Theory
Three atoms are placed in an otherwise empty system. The relative positions of the atoms are determined by the following three coordinates
r_ij is the radial distance between atoms i and j,
r_ik is the radial distance between atoms i and k, and
theta_ijk is the angle formed between the i-j and i-k vectors.
Based on these three bond coordinates, the full positions of the three atoms in the system are determined as follows
Atom i is positioned at the system’s origin, [0, 0, 0]
Atom j is placed r_ij away from atom i along the x coordinate, [r_ij, 0.0, 0.0]
Atom k is placed in the xy plane based on r_ik and theta_ijk, [r_ik cos(theta_ijk), r_ik sin(theta_ijk), 0.0]
Values of r_ij, r_ik and theta_ijk are iterated over. The potential energy of the three atoms is evaluated for each configuration corresponding to the different coordinate sets.
2. Define calculation functions and generate files
This section defines the calculation functions and associated resource files exactly as they exist inside the iprPy package. This allows for the code used to be directly visible and modifiable by anyone looking to see how it works.
2.1. bond_angle_scan()
This is the primary function for the calculation. The version of this function built in iprPy can be accessed by calling the calc() method of an object of the associated calculation class.
[4]:
def bond_angle_scan(lammps_command: str,
potential: am.lammps.Potential,
symbols: list,
mpi_command: Optional[str] = None,
rmin: float = uc.set_in_units(0.5, 'angstrom'),
rmax: float = uc.set_in_units(6.0, 'angstrom'),
rnum: int = 100,
thetamin: float = 1.0,
thetamax: float = 180,
thetanum: int = 100) -> dict:
"""
Performs a three-body bond angle energy scan over a range of interatomic
spaces, r, and angles, theta.
Parameters
----------
lammps_command :str
Command for running LAMMPS.
potential : atomman.lammps.Potential
The LAMMPS implemented potential to use.
symbols : list
The potential symbols associated with the three atoms in the cluster.
mpi_command : str, optional
The MPI command for running LAMMPS in parallel. If not given, LAMMPS
will run serially.
rmin : float, optional
The minimum value for the r_ij and r_ik spacings. Default value is 0.5.
rmax : float, optional
The maximum value for the r_ij and r_ik spacings. Default value is 5.5.
rnum : int, optional
The number of r_ij and r_ik spacings to evaluate. Default value is 100.
thetamin : float, optional
The minimum value for the theta angle. Default value is 1.0.
thetamax : float, optional
The maximum value for the theta angle. Default value is 180.0.
thetanum : int, optional
The number of theta angles to evaluate. Default value is 100.
Returns
-------
dict
Dictionary of results consisting of keys:
- **'cluster'** (*atomman.cluster.BondAngleMap*) - Object that maps
measured energies to r, theta coordinates, and contains built-in
analysis tools.
- **results_file'** (*str*) - File name containing the raw energy
scan results.
- **'length_unit'** (*str*) - Unit of length used in the results_file.
- **'energy_unit'** (*str*) - Unit of energy used in the results_file.
"""
# Create cluster object
cluster = am.cluster.BondAngleMap(rmin=rmin, rmax=rmax, rnum=rnum,
thetamin=thetamin, thetamax=thetamax,
thetanum=thetanum, symbols=symbols)
# Get lammps units
lammps_units = lmp.style.unit(potential.units)
# Define lammps variables
lammps_variables = {}
# Add range parameters
lammps_variables['rmin'] = uc.get_in_units(rmin, lammps_units['length'])
lammps_variables['rmax'] = uc.get_in_units(rmax, lammps_units['length'])
lammps_variables['rnum'] = uc.get_in_units(rnum, lammps_units['length'])
lammps_variables['thetamin'] = thetamin
lammps_variables['thetamax'] = thetamax
lammps_variables['thetanum'] = thetanum
# Add atomic types
if len(cluster.symbols) == 1:
natypes = 1
atype = np.array([1,1,1])
symbols = cluster.symbols
elif len(cluster.symbols) == 3:
symbols, atype = np.unique(cluster.symbols, return_inverse=True)
atype += 1
natypes = len(symbols)
lammps_variables['natypes'] = natypes
lammps_variables['atype1'] = atype[0]
lammps_variables['atype2'] = atype[1]
lammps_variables['atype3'] = atype[2]
# Add potential information
lammps_variables['atomman_pair_info'] = potential.pair_info(symbols)
lammps_variables['atom_style'] = potential.atom_style
lammps_variables['units'] = potential.units
# Build lammps input script
lammps_script = 'bond_scan.in'
template = read_calc_file('iprPy.calculation.bond_angle_scan', 'bond_scan.template')
with open(lammps_script, 'w') as f:
f.write(filltemplate(template, lammps_variables, '<', '>'))
# Run lammps and extract data
lmp.run(lammps_command, script_name=lammps_script, mpi_command=mpi_command, logfile=None, screen=False)
cluster.load_table('3_body_scan.txt', length_unit=lammps_units['length'],
energy_unit=lammps_units['energy'])
# Collect results
results_dict = {}
results_dict['cluster'] = cluster
results_dict['results_file'] = '3_body_scan.txt'
results_dict['length_unit'] = lammps_units['length']
results_dict['energy_unit'] = lammps_units['energy']
return results_dict
2.2. bond_scan.template file
[5]:
with open('bond_scan.template', 'w') as f:
f.write("""# LAMMPS input script for exploring the three-body phase space
# Specify loop ranges
variable rmin equal <rmin>
variable rmax equal <rmax>
variable rnum equal <rnum>
variable thetamin equal <thetamin>
variable thetamax equal <thetamax>
variable thetanum equal <thetanum>
# Define dummy values for loop indices
#variable i equal 1
#variable j equal 1
#variable k equal 1
# Define variable atom coordinates
variable rij equal ${rmin}+(v_i-1)*(${rmax}-${rmin})/(${rnum}-1)
variable rik equal ${rmin}+(v_j-1)*(${rmax}-${rmin})/(${rnum}-1)
variable theta equal ${thetamin}+(v_k-1)*(${thetamax}-${thetamin})/(${thetanum}-1)
variable rtheta equal v_theta*PI/180.0
variable j_x equal v_rij
variable k_x equal v_rik*cos(v_rtheta)
variable k_y equal v_rik*sin(v_rtheta)
variable energy equal pe
# Define box bounds based on rmax
variable rlo equal -3*${rmax}
variable rhi equal 3*${rmax}
units <units>
atom_style <atom_style>
boundary f f f
region rbox block ${rlo} ${rhi} ${rlo} ${rhi} -1.0 1.0
create_box <natypes> rbox
<atomman_pair_info>
thermo_style custom step pe
thermo_modify format float %.13e
# Create atom 1
create_atoms <atype1> single 0.0 0.0 0.0 units box
# Start 3_body_scan.txt with header fields
print "${rmin} ${rmax} ${rnum}" file 3_body_scan.txt screen no
print "${rmin} ${rmax} ${rnum}" append 3_body_scan.txt screen no
print "${thetamin} ${thetamax} ${thetanum}" append 3_body_scan.txt screen no
# Loop i over r_ij values
variable i loop ${rnum}
label loopi
# Create atom 2
create_atoms <atype2> single ${j_x} 0.0 0.0 units box
group id2 id 2
# Loop j over r_ik values
variable j loop ${rnum}
label loopj
# Loop k over theta values
variable k loop ${thetanum}
label loopk
# Create atom 3
create_atoms <atype3> single ${k_x} ${k_y} 0.0 units box
group id3 id 3
# Compute energy and output it to 3_body_scan.txt
run 0
print "${i} ${j} ${k} ${energy}" append 3_body_scan.txt screen no
delete_atoms group id3
next k
jump bond_scan.in loopk
next j
jump bond_scan.in loopj
delete_atoms group id2
next i
jump bond_scan.in loopi""")
3. Specify input parameters
3.1. System-specific paths
lammps_command is the LAMMPS command to use (required).
mpi_command MPI command for running LAMMPS in parallel. A value of None will run simulations serially.
[6]:
lammps_command = 'lmp'
mpi_command = None
# Optional: check that LAMMPS works and show its version
print(f'LAMMPS version = {am.lammps.checkversion(lammps_command)["version"]}')
LAMMPS version = 15 Sep 2022
3.2. Interatomic potential
potential_name gives the name of the potential_LAMMPS reference record in the iprPy library to use for the calculation.
potential is an atomman.lammps.Potential object (required).
[7]:
potential_name = '1999--Mishin-Y--Ni--LAMMPS--ipr1'
# Retrieve potential and parameter file(s) using atomman
potential = am.load_lammps_potential(id=potential_name, getfiles=True)
2.3. Calculation-specific parameters
symbols is the element or set of three element model symbols to use for the cluster.
rmin is the minimum r_ij and r_ik spacing to use.
rmax is the maximum r_ij and r_ik spacing to use.
rnum is the number of r_ij and r_ik spacing steps to evaluate.
thetamin is the minimum theta angle to use.
thetamax is the maximum theta angle to use.
thetanum is the number of theta angle steps to evaluate.
[8]:
symbols = 'Ni'
rmin = uc.set_in_units(0.02, 'angstrom')
rmax = uc.set_in_units(6.0, 'angstrom')
rnum = 20
thetamin = 1.0
thetamax = 180.0
thetanum = 20
4. Run calculation and view results
4.1. Run calculation
All primary calculation method functions take a series of inputs and return a dictionary of outputs.
[9]:
results_dict = bond_angle_scan(lammps_command, potential, symbols, mpi_command = mpi_command,
rmin = rmin, rmax = rmax, rnum = rnum,
thetamin = thetamin, thetamax = thetamax, thetanum = thetanum)
print(results_dict.keys())
dict_keys(['cluster', 'results_file', 'length_unit', 'energy_unit'])
4.2. Report results
Values returned in the results_dict: - ‘cluster’ (atomman.cluster.BondAngleMap) - An object containing the evaluated energies and their coordinates. Includes built-in methods for analyzing the results. - ‘results_file’ (str) - The name of the file where the measured energies are saved. - ‘length_unit’ (str) - The unit of length used in results_file. - ‘energy_unit’ (str) - The unit of energy used in results_file.
[10]:
print('Results are saved in', results_dict['results_file'])
print('with lengths in', results_dict['length_unit'])
print('and energies in', results_dict['energy_unit'])
Results are saved in 3_body_scan.txt
with lengths in angstrom
and energies in eV
[11]:
cluster = results_dict['cluster']
cluster.df
[11]:
| r_ij | r_ik | r_jk | theta | energy | |
|---|---|---|---|---|---|
| 0 | 0.02 | 0.02 | 0.000349 | 1.000000 | 9.662228e+02 |
| 1 | 0.02 | 0.02 | 0.003633 | 10.421053 | 9.644323e+02 |
| 2 | 0.02 | 0.02 | 0.006892 | 19.842105 | 9.626081e+02 |
| 3 | 0.02 | 0.02 | 0.010104 | 29.263158 | 9.608158e+02 |
| 4 | 0.02 | 0.02 | 0.013248 | 38.684211 | 9.590673e+02 |
| ... | ... | ... | ... | ... | ... |
| 7995 | 6.00 | 6.00 | 11.356946 | 142.315789 | -9.291009e-11 |
| 7996 | 6.00 | 6.00 | 11.636851 | 151.736842 | -9.291009e-11 |
| 7997 | 6.00 | 6.00 | 11.838145 | 161.157895 | -9.291009e-11 |
| 7998 | 6.00 | 6.00 | 11.959468 | 170.578947 | -9.291009e-11 |
| 7999 | 6.00 | 6.00 | 12.000000 | 180.000000 | -9.291009e-11 |
8000 rows × 5 columns
[12]:
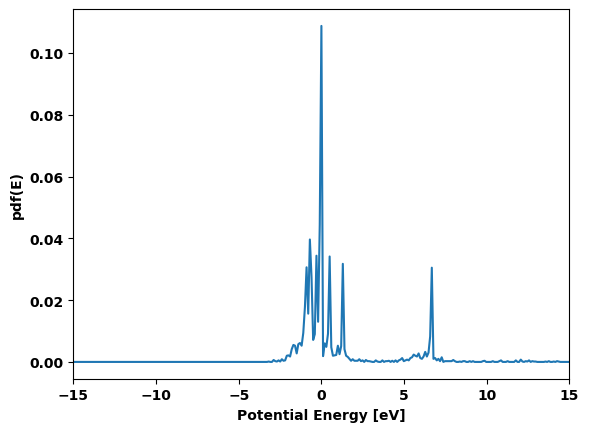
cluster.plot_pdf()
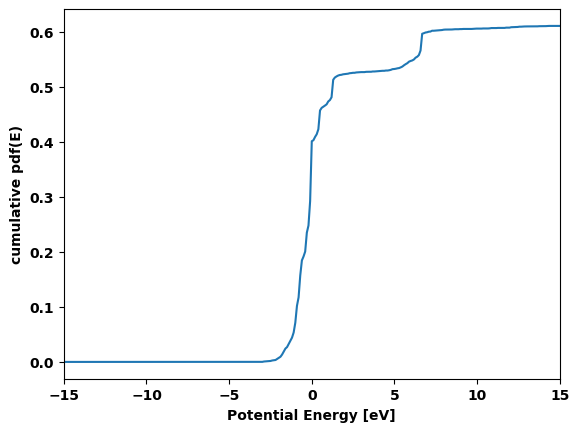
cluster.plot_cumulative_pdf()
None