-
Citation: X.W. Zhou, N.C. Bartelt, and R.B. Sills (2021), "Enabling simulations of helium bubble nucleation and growth: A strategy for interatomic potentials", Physical Review B 103(1), 014108. DOI: 10.1103/physrevb.103.014108.Abstract: Helium bubbles are a severe form of radiation damage that has been frequently observed. It would be possible to understand the complex processes that cause bubble formation if suitable interatomic potentials were available to enable molecular dynamics simulations. In this paper, Pd-H-He embedded-atom method potentials based on both Daw-Baskes and Finnis-Sinclair formalisms have been developed to enable modeling of He bubbles formed by the radioactive decay of tritium in Pd. Our potentials incorporate helium into an existing Pd-H potential while addressing two challenging paradoxes: (a) Interstitial He atoms can dramatically lower their energies by forming dimers and larger clusters in Pd but are only bound by weak van der Waals forces in the gas phase. (b) He atoms diffuse readily in Pd yet significantly distort the Pd lattice with large volume expansions. We demonstrate that both of our potentials reproduce density functional theory results for (b). However, the Daw-Baskes formalism fails to resolve paradox (a) because it cannot reproduce the experimental helium equation of state. We resolved this problem through a modification of the Finnis-Sinclair formalism in which a (fictitious) negative embedding charge density is produced by Pd at the He binding sites. In addition to molecular statics validation of static properties, molecular dynamics simulation tests establish that our Finnis-Sinclair potential leads to the nucleation of helium bubbles from an initial random distribution of helium interstitial atoms.
-
LAMMPS pair_style eam/he (2021--Zhou-X-W--Pd-H-He--LAMMPS--ipr1)See Computed Properties
Notes: This file was provided by Xiaowang Zhou (Sandia) on March 24, 2021 and posted with his permission. The eam/he pair style was added to LAMMPS starting with the 10 Feb 2021 version.
File(s):
Implementation Information
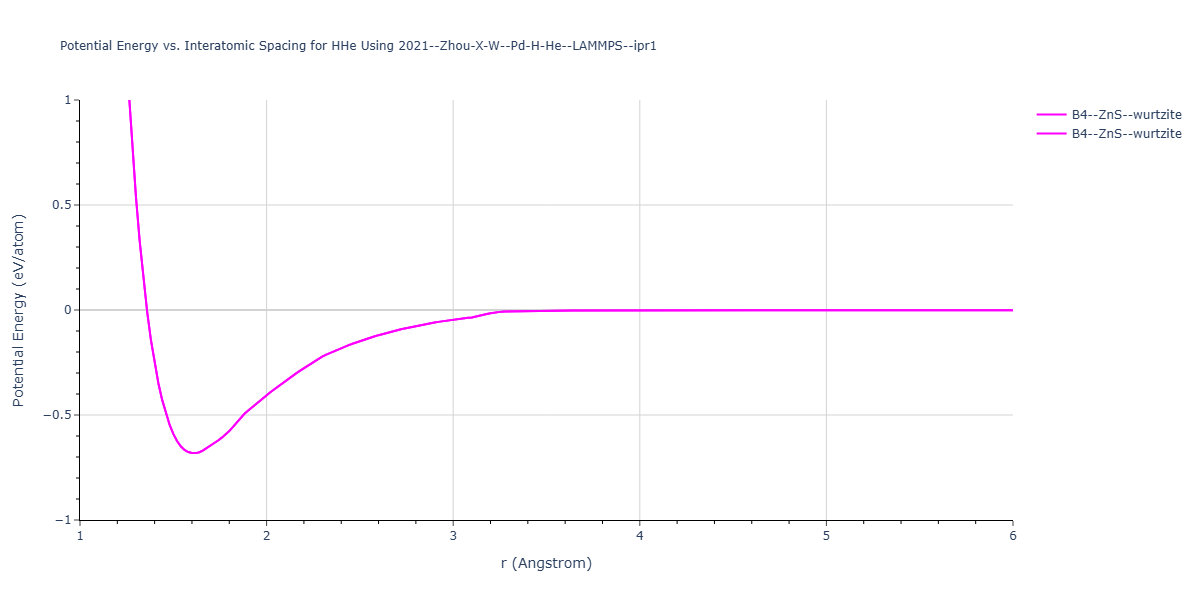
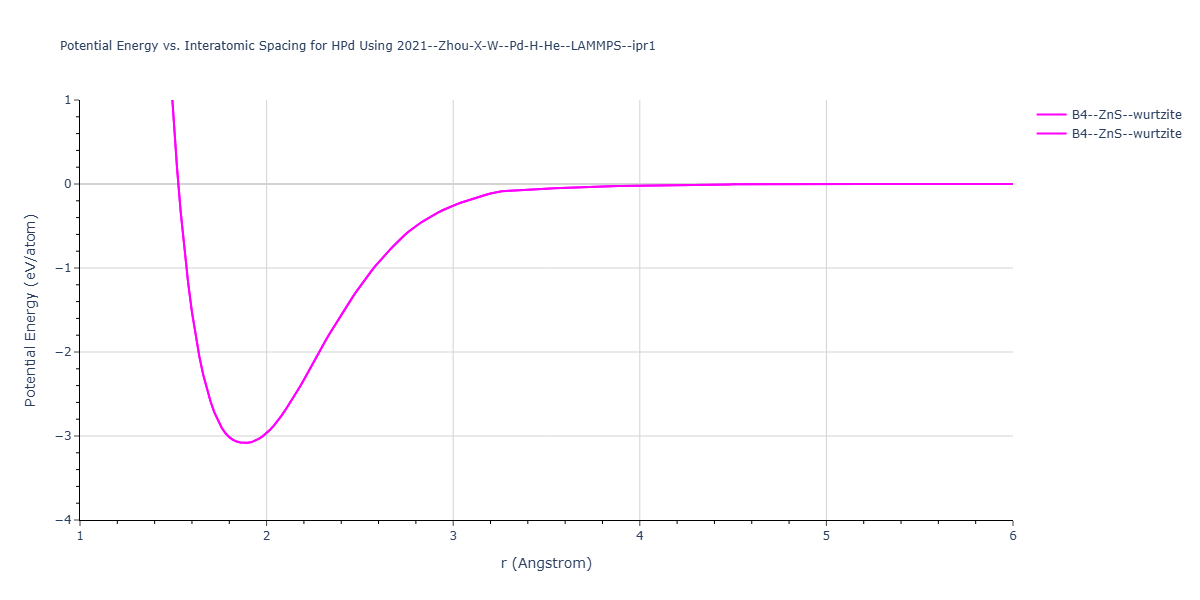
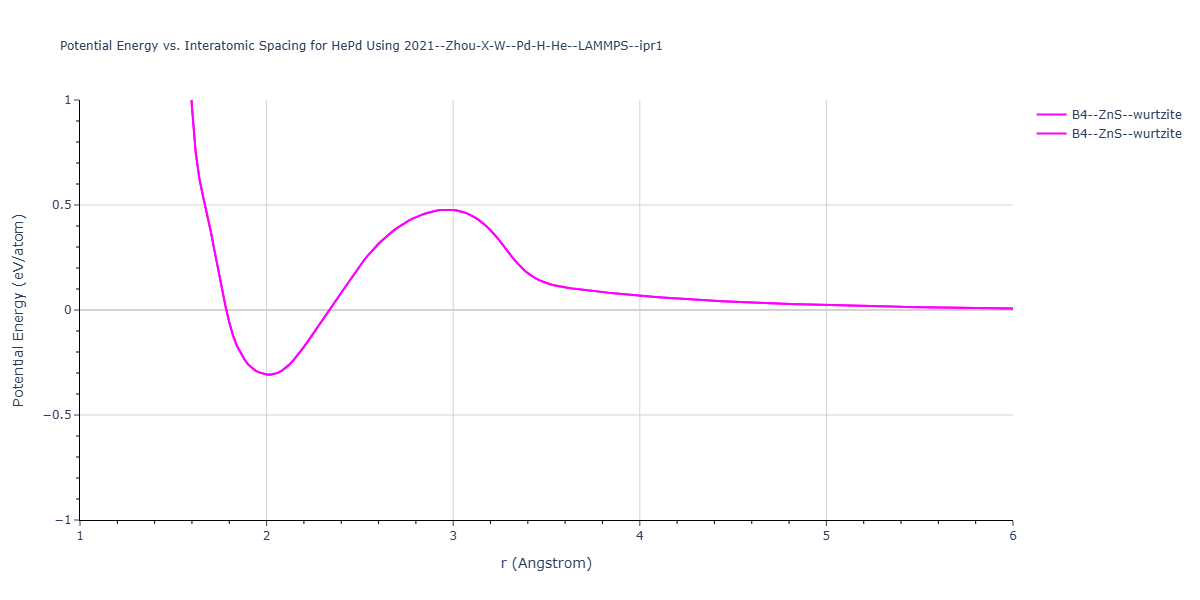
Cohesive Energy vs. Interatomic Spacing
Plots of potential energy vs interatomic spacing, r, are shown below for a number of crystal structures. The structures are generated based on the ideal atomic positions and b/a and c/a lattice parameter ratios for a given crystal prototype. The size of the system is then uniformly scaled, and the energy calculated without relaxing the system. To obtain these plots, values of r are evaluated every 0.02 Å up to 6 Å.
The calculation method used is available as the iprPy E_vs_r_scan calculation method.
Clicking on the image of a plot will open an interactive version of it in a new tab. The underlying data for the plots can be downloaded by clicking on the links above each plot.
Notes and Disclaimers:
- These values are meant to be guidelines for comparing potentials, not the absolute values for any potential's properties. Values listed here may change if the calculation methods are updated due to improvements/corrections. Variations in the values may occur for variations in calculation methods, simulation software and implementations of the interatomic potentials.
- The minima identified by this calculation do not guarantee that the associated crystal structures will be stable since no relaxation is performed.
- NIST disclaimer
Version Information:
- 2020-12-18. Descriptions, tables and plots updated to reflect that the energy values are the measuredper atom potential energy rather than cohesive energy as some potentials have non-zero isolated atom energies.
- 2019-02-04. Values regenerated with even r spacings of 0.02 Å, and now include values less than 2 Å when possible. Updated calculation method and parameters enhance compatibility with more potential styles.
- 2019-04-26. Results for hcp, double hcp, α-As and L10 prototypes regenerated from different unit cell representations. Only α-As results show noticable (>1e-5 eV) difference due to using a different coordinate for Wykoff site c position.
- 2018-06-13. Values for MEAM potentials corrected. Dynamic versions of the plots moved to separate pages to improve page loading. Cosmetic changes to how data is shown and updates to the documentation.
- 2017-01-11. Replaced png pictures with interactive Bokeh plots. Data regenerated with 200 values of r instead of 300.
- 2016-09-28. Plots for binary structures added. Data and plots for elemental structures regenerated. Data values match the values of the previous version. Data table formatting slightly changed to increase precision and ensure spaces between large values. Composition added to plot title and structure names made longer.
- 2016-04-07. Plots for elemental structures added.