Warning! Note that elemental potentials taken from alloy descriptions may not work well for the pure species. This is particularly true if the elements were fit for compounds instead of being optimized separately. As with all interatomic potentials, please check to make sure that the performance is adequate for your problem.
Citation: Y. Mishin, M.J. Mehl, and D.A. Papaconstantopoulos (2005), "Phase stability in the Fe-Ni system: Investigation by first-principles calculations and atomistic simulations", Acta Materialia, 53(15), 4029-4041. DOI: 10.1016/j.actamat.2005.05.001.
Abstract: First-principles calculations of the energy of various crystal structures of Fe, Ni and ordered Fe–Ni compounds with different stoichiometries have been performed by the linearized augmented plane wave (LAPW) method in the generalized gradient approximation. The most stable compounds are L12–Ni3Fe, L10–FeNi, C11f–Ni2Fe and C11f–Fe2Ni. The L12-Ni3Fe compound has the largest negative formation energy, which is consistent with the experimental Fe–Ni phase diagram. The L10–FeNi compound has also been observed experimentally in meteorite samples as a metastable phase. It is suggested here that the C11f compounds could also form in Fe–Ni alloys at low temperatures. A new semi-empirical interatomic potential has been developed for the Fe–Ni system by fitting to experimental data and the results of the LAPW calculations. Recognizing the significance of the covalent component of bonding in this system, the potential is based on the embedded-atom method (EAM) but additionally includes a bond-angle dependence. In comparison with the existing modified EAM method, our potential form is simpler, extends interactions to several (3–5) coordination shells and replaces the screening procedure by a smooth cutoff of the potential functions. The potential reproduces a variety of properties of Fe and Ni with a reasonable accuracy. It also reproduces all stability trends across the Fe–Ni system established by the LAPW calculations. The potential can be useful in atomistic simulations of the phases of the Fe–Ni system.
Notes: These files were provided by Yuri Mishin (George Mason University) and posted on 22 Dec. 2009. Prof. Mishin requested the following note be included: "The equation appearing in the Appendix on page 4040 contains a typing error: the sign before 1/3 in the last line must be negative." He provided the corrected equation for the angular-dependent force contributions in ADP_Forces.jpg or ADP_Forces.pdf. File(s):
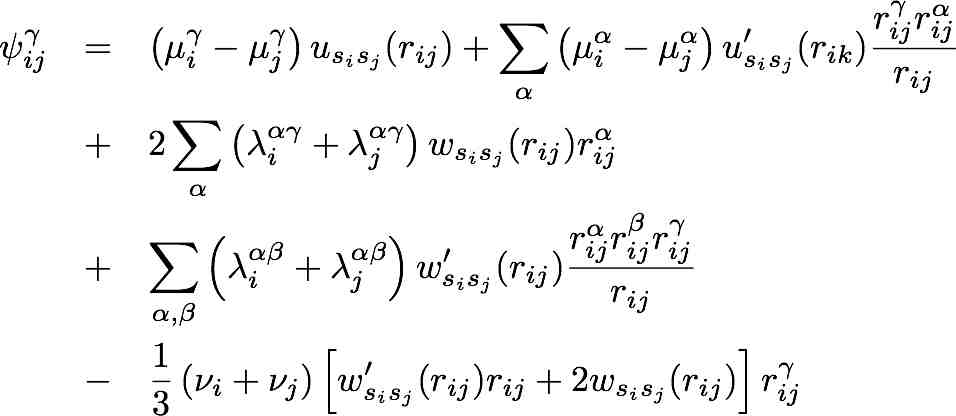{kind=link}