Introduction to atomman: LAMMPS functionality
Lucas M. Hale, lucas.hale@nist.gov, Materials Science and Engineering Division, NIST.
1. Introduction
This Notebook provides an introduction to interacting with LAMMPS using atomman by working through a demonstration simulation.
Library Imports
[1]:
# Standard libraries
from pathlib import Path
import datetime
# http://www.numpy.org/
import numpy as np
# http://matplotlib.org/
import matplotlib.pyplot as plt
%matplotlib inline
# https://github.com/usnistgov/atomman
import atomman as am
import atomman.lammps as lmp
import atomman.unitconvert as uc
# Show atomman version
print('atomman version =', am.__version__)
# Show date of Notebook execution
print('Notebook executed on', datetime.date.today())
atomman version = 1.4.11
Notebook executed on 2024-04-29
2. Interatomic Potential Control
LAMMPS is capable of using a wide array of interatomic potential styles, which are defined through a combination of LAMMPS commands and potential parameter files. In atomman, the LAMMPS commands can be automatically generated using the Potentials class and structured data model files.
2.1. Load potential from database
Any LAMMPS-compatible potential in the NIST Interatomic Potentials Repository can be loaded/downloaded using load_lammps_potential().
[2]:
# Load potential based on its unique ID
pot_id = '2015--Pascuet-M-I--Al--LAMMPS--ipr1'
potential = am.load_lammps_potential(id=pot_id, getfiles=True)
2.2. Potential parameter files
Setting getfiles=True will copy/download the potential parameter files from the local/remote library database to the current working directory in a subfolder matching the potential’s id.
[3]:
for fname in Path(pot_id).glob('*'):
print(fname)
with open(fname) as f:
print(f.read())
print()
2.3. lammps.Potential Class
The load_lammps_potential() function returns a Potential object that can be used to explore properties of the potential and generate LAMMPS input commands.
Basic properties can be directly obtained.
[4]:
# Show that basic properties can be directly obtained
print('str(potential) -> ', potential)
print('potential.units -> ', potential.units)
print('potential.atom_style -> ', potential.atom_style)
print('potential.pair_style -> ', potential.pair_style)
print('potential.symbols -> ', potential.symbols)
print('potential.elements() -> ', potential.elements())
print('potential.masses() -> ', potential.masses())
str(potential) -> potential_LAMMPS record named 2015--Pascuet-M-I--Al--LAMMPS--ipr1
potential.units -> metal
potential.atom_style -> atomic
potential.pair_style -> meam
potential.symbols -> ['Al']
potential.elements() -> ['Al']
potential.masses() -> [26.9815]
The LAMMPS command lines for the potential are auto-generated based on a list of symbols corresponding to LAMMPS atom types. This fully works for all LAMMPS pair_styles, with only the hybrid and hybrid/overlay styles having limitations.
[5]:
print('potential.pair_info() ->')
print(potential.pair_info())
potential.pair_info() ->
print "Potential 2015--Pascuet-M-I--Al--LAMMPS--ipr1 listed in the NIST Interatomic Potentials Repository:"
print "https://www.ctcms.nist.gov/potentials/entry/2015--Pascuet-M-I-Fernandez-J-R--Al/2015--Pascuet-M-I--Al--LAMMPS--ipr1.html"
print "Publication(s) related to the potential:"
print "https://doi.org/10.1016/j.jnucmat.2015.09.030"
print "Parameter file(s) can be downloaded at:"
print "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/Al.meam"
print "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/library-Al.meam"
pair_style meam
pair_coeff * * library-Al.meam Al Al.meam Al
mass 1 26.9815
[6]:
print("potential.pair_info(['Al', 'Al', 'Al']) ->")
print(potential.pair_info(['Al', 'Al', 'Al']))
potential.pair_info(['Al', 'Al', 'Al']) ->
print "Potential 2015--Pascuet-M-I--Al--LAMMPS--ipr1 listed in the NIST Interatomic Potentials Repository:"
print "https://www.ctcms.nist.gov/potentials/entry/2015--Pascuet-M-I-Fernandez-J-R--Al/2015--Pascuet-M-I--Al--LAMMPS--ipr1.html"
print "Publication(s) related to the potential:"
print "https://doi.org/10.1016/j.jnucmat.2015.09.030"
print "Parameter file(s) can be downloaded at:"
print "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/Al.meam"
print "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/library-Al.meam"
pair_style meam
pair_coeff * * library-Al.meam Al Al.meam Al Al Al
mass 1 26.9815
mass 2 26.9815
mass 3 26.9815
3. Generate initial system
3.1. Load relaxed crystal
A crystalline system can be easily generated using a unit cell system either defined in atomman, imported from another format, or accessed from the potentials database. For simplicity, here we will get the relaxed fcc structure for the potential.
Load fcc prototype with Al lattice constant
[7]:
system = am.load('crystal', family='A1--Cu--fcc', potential=potential)
print(system)
avect = [ 4.050, 0.000, 0.000]
bvect = [ 0.000, 4.050, 0.000]
cvect = [ 0.000, 0.000, 4.050]
origin = [ 0.000, 0.000, 0.000]
natoms = 4
natypes = 1
symbols = ('Al',)
pbc = [ True True True]
per-atom properties = ['atype', 'pos']
id atype pos[0] pos[1] pos[2]
0 1 0.000 0.000 0.000
1 1 0.000 2.025 2.025
2 1 2.025 0.000 2.025
3 1 2.025 2.025 0.000
3.2. Manipulate system
More complicated atomic configurations can then be generated by manipulating the seed system and the atoms contained within. Here, we’ll limit the manipulations to making the system a 3x3x3 supercell of itself.
[8]:
system = system.supersize(3,3,3)
print('Supercell has', system.natoms, 'atoms')
Supercell has 108 atoms
3.3. Save to atom data file
System.dump(‘atom_data’) outputs the system to a LAMMPS atom data file. Quick notes on the parameters used here
Giving potential allows for the appropriate units and atom_style settings to be used.
The float_format value used here is a small precision to enhance clarity of print statements below. Typically, the precision should be large (default value is ‘%.13f’).
Setting return_pair_info=True will return the LAMMPS commands for both the system and the potential together. This is the preferred method as it ensures compatibility with all potential pair styles currently in the database.
[9]:
# Save System to 'atom.dat' atom data file
system_pair_info = system.dump('atom_data', f='atom.dat',
potential=potential,
float_format='%.4f', # Remove or make larger precision for real runs!
)
Show the returned LAMMPS command lines
[10]:
print(system_pair_info)
units metal
atom_style atomic
boundary p p p
read_data atom.dat
print "Potential 2015--Pascuet-M-I--Al--LAMMPS--ipr1 listed in the NIST Interatomic Potentials Repository:"
print "https://www.ctcms.nist.gov/potentials/entry/2015--Pascuet-M-I-Fernandez-J-R--Al/2015--Pascuet-M-I--Al--LAMMPS--ipr1.html"
print "Publication(s) related to the potential:"
print "https://doi.org/10.1016/j.jnucmat.2015.09.030"
print "Parameter file(s) can be downloaded at:"
print "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/Al.meam"
print "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/library-Al.meam"
pair_style meam
pair_coeff * * library-Al.meam Al Al.meam Al
mass 1 26.9815
Show the contents of the data file
[11]:
with open('atom.dat') as f:
print(f.read())
108 atoms
1 atom types
0.0000 12.1500 xlo xhi
0.0000 12.1500 ylo yhi
0.0000 12.1500 zlo zhi
Atoms # atomic
1 1 0.0000 0.0000 0.0000
2 1 0.0000 2.0250 2.0250
3 1 2.0250 0.0000 2.0250
4 1 2.0250 2.0250 0.0000
5 1 4.0500 0.0000 0.0000
6 1 4.0500 2.0250 2.0250
7 1 6.0750 0.0000 2.0250
8 1 6.0750 2.0250 0.0000
9 1 8.1000 0.0000 0.0000
10 1 8.1000 2.0250 2.0250
11 1 10.1250 0.0000 2.0250
12 1 10.1250 2.0250 0.0000
13 1 0.0000 4.0500 0.0000
14 1 0.0000 6.0750 2.0250
15 1 2.0250 4.0500 2.0250
16 1 2.0250 6.0750 0.0000
17 1 4.0500 4.0500 0.0000
18 1 4.0500 6.0750 2.0250
19 1 6.0750 4.0500 2.0250
20 1 6.0750 6.0750 0.0000
21 1 8.1000 4.0500 0.0000
22 1 8.1000 6.0750 2.0250
23 1 10.1250 4.0500 2.0250
24 1 10.1250 6.0750 0.0000
25 1 0.0000 8.1000 0.0000
26 1 0.0000 10.1250 2.0250
27 1 2.0250 8.1000 2.0250
28 1 2.0250 10.1250 0.0000
29 1 4.0500 8.1000 0.0000
30 1 4.0500 10.1250 2.0250
31 1 6.0750 8.1000 2.0250
32 1 6.0750 10.1250 0.0000
33 1 8.1000 8.1000 0.0000
34 1 8.1000 10.1250 2.0250
35 1 10.1250 8.1000 2.0250
36 1 10.1250 10.1250 0.0000
37 1 0.0000 0.0000 4.0500
38 1 0.0000 2.0250 6.0750
39 1 2.0250 0.0000 6.0750
40 1 2.0250 2.0250 4.0500
41 1 4.0500 0.0000 4.0500
42 1 4.0500 2.0250 6.0750
43 1 6.0750 0.0000 6.0750
44 1 6.0750 2.0250 4.0500
45 1 8.1000 0.0000 4.0500
46 1 8.1000 2.0250 6.0750
47 1 10.1250 0.0000 6.0750
48 1 10.1250 2.0250 4.0500
49 1 0.0000 4.0500 4.0500
50 1 0.0000 6.0750 6.0750
51 1 2.0250 4.0500 6.0750
52 1 2.0250 6.0750 4.0500
53 1 4.0500 4.0500 4.0500
54 1 4.0500 6.0750 6.0750
55 1 6.0750 4.0500 6.0750
56 1 6.0750 6.0750 4.0500
57 1 8.1000 4.0500 4.0500
58 1 8.1000 6.0750 6.0750
59 1 10.1250 4.0500 6.0750
60 1 10.1250 6.0750 4.0500
61 1 0.0000 8.1000 4.0500
62 1 0.0000 10.1250 6.0750
63 1 2.0250 8.1000 6.0750
64 1 2.0250 10.1250 4.0500
65 1 4.0500 8.1000 4.0500
66 1 4.0500 10.1250 6.0750
67 1 6.0750 8.1000 6.0750
68 1 6.0750 10.1250 4.0500
69 1 8.1000 8.1000 4.0500
70 1 8.1000 10.1250 6.0750
71 1 10.1250 8.1000 6.0750
72 1 10.1250 10.1250 4.0500
73 1 0.0000 0.0000 8.1000
74 1 0.0000 2.0250 10.1250
75 1 2.0250 0.0000 10.1250
76 1 2.0250 2.0250 8.1000
77 1 4.0500 0.0000 8.1000
78 1 4.0500 2.0250 10.1250
79 1 6.0750 0.0000 10.1250
80 1 6.0750 2.0250 8.1000
81 1 8.1000 0.0000 8.1000
82 1 8.1000 2.0250 10.1250
83 1 10.1250 0.0000 10.1250
84 1 10.1250 2.0250 8.1000
85 1 0.0000 4.0500 8.1000
86 1 0.0000 6.0750 10.1250
87 1 2.0250 4.0500 10.1250
88 1 2.0250 6.0750 8.1000
89 1 4.0500 4.0500 8.1000
90 1 4.0500 6.0750 10.1250
91 1 6.0750 4.0500 10.1250
92 1 6.0750 6.0750 8.1000
93 1 8.1000 4.0500 8.1000
94 1 8.1000 6.0750 10.1250
95 1 10.1250 4.0500 10.1250
96 1 10.1250 6.0750 8.1000
97 1 0.0000 8.1000 8.1000
98 1 0.0000 10.1250 10.1250
99 1 2.0250 8.1000 10.1250
100 1 2.0250 10.1250 8.1000
101 1 4.0500 8.1000 8.1000
102 1 4.0500 10.1250 10.1250
103 1 6.0750 8.1000 10.1250
104 1 6.0750 10.1250 8.1000
105 1 8.1000 8.1000 8.1000
106 1 8.1000 10.1250 10.1250
107 1 10.1250 8.1000 10.1250
108 1 10.1250 10.1250 8.1000
4. Converting to/from LAMMPS units
LAMMPS performs its calculations with values in one of multiple sets of pre-defined units. The atomman.lammps.style submodule has some useful functions when working with different units options.
atomman.lammps.style.timestep()
The lammps.style.timestep() function returns the default timestep value for a given LAMMPS units option.
Parameters
units (str) the LAMMPS units option being used.
atomman.lammps.style.unit()
The lammps.style.unit() function returns a dictionary giving the units associated with physical quantities as used by LAMMPS with a given units option.
Parameters
units (str) the LAMMPS units option being used.
[12]:
timestep = lmp.style.timestep(potential.units)
print(timestep)
0.001
[13]:
lammps_unit = lmp.style.unit(potential.units)
print(lammps_unit)
OrderedDict([('mass', 'g/mol'), ('length', 'angstrom'), ('time', 'ps'), ('energy', 'eV'), ('velocity', 'angstrom/ps'), ('force', 'eV/angstrom'), ('torque', 'eV'), ('temperature', 'K'), ('pressure', 'bar'), ('dynamic viscosity', 'Pa*s/10'), ('charge', 'e'), ('dipole', 'e*angstrom'), ('electric field', 'V/angstrom'), ('density', 'g/cm^3'), ('ang-mom', 'angstrom*angstrom/ps*g/mol'), ('ang-vel', '1/ps')])
5. Composing LAMMPS Input Scripts
LAMMPS scripts can easily be constructed by combining the system_info generated from System.dump(‘atom_data’), the pair_info from Potential.pair_info(), and any user-defined input lines. This allows for specific simulation actions to easily be perfored across different potentials or initial configurations.
5.1. Show content generated in previous sections
Show system_pair_info generated in Section 3.3.
[14]:
print(system_pair_info)
units metal
atom_style atomic
boundary p p p
read_data atom.dat
print "Potential 2015--Pascuet-M-I--Al--LAMMPS--ipr1 listed in the NIST Interatomic Potentials Repository:"
print "https://www.ctcms.nist.gov/potentials/entry/2015--Pascuet-M-I-Fernandez-J-R--Al/2015--Pascuet-M-I--Al--LAMMPS--ipr1.html"
print "Publication(s) related to the potential:"
print "https://doi.org/10.1016/j.jnucmat.2015.09.030"
print "Parameter file(s) can be downloaded at:"
print "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/Al.meam"
print "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/library-Al.meam"
pair_style meam
pair_coeff * * library-Al.meam Al Al.meam Al
mass 1 26.9815
5.2. Write LAMMPS input script template
LAMMPS scripts can be dynamically generated using Python functions or templates that take the above info lines and other values as parameters. Here, we demonstrate a LAMMPS input template script where all fields to be filled in in Python are delimited with <> brackets.
[15]:
template = """
<system_pair_info>
# Define temperature and dependent variables
variable T equal <temperature>
variable twoT equal 2*$T
# Define equilibrium pressure
variable P equal <pressure>
# Define timestep and dependent variables
variable deltat equal <timestep>
variable Trelax equal 100*${deltat}
variable Prelax equal 1000*${deltat}
# Initialize atomic velocities with twoT
velocity all create ${twoT} 124352
# Define thermo steps and properties
thermo 100
thermo_style custom step temp press lx ly lz
# Define dump
dump mydump all atom 100000 *.dump
# Specify timestep
timestep ${deltat}
# Apply npt conditions
fix 1 all npt temp $T $T ${Trelax} iso $P $P ${Prelax}
# Run simulation
run 100000
"""
5.3. Fill in the template with atomman.tools.filltemplate()
The template can then be easily filled in with the atomman.tools.filltemplate() function.
Parameters
template (str or file-like object) is the template to fill in.
variable (dict) gives the delimited keys and corresponding values to insert into the template.
s_delimiter (str) the starting delimiter for identifying variable names.
e_delimiter (str) the ending delimiter for identifying variable names.
Build dictionary of template variables
[16]:
lammps_variable = {}
# Generated above
lammps_variable['system_pair_info'] = system_pair_info
# Set timestep to default value for LAMMPS units
lammps_variable['timestep'] = lmp.style.timestep(units=potential.units)
# Specify temperature to equilibriate at (always in Kelvin)
lammps_variable['temperature'] = 100
# Specify pressure to equilibriate at
pressure = uc.set_in_units(0.0, 'MPa')
lammps_variable['pressure'] = uc.get_in_units(pressure, lammps_unit['pressure'])
print(lammps_variable)
{'system_pair_info': 'units metal\natom_style atomic\n\nboundary p p p\nread_data atom.dat\n\nprint "Potential 2015--Pascuet-M-I--Al--LAMMPS--ipr1 listed in the NIST Interatomic Potentials Repository:"\nprint "https://www.ctcms.nist.gov/potentials/entry/2015--Pascuet-M-I-Fernandez-J-R--Al/2015--Pascuet-M-I--Al--LAMMPS--ipr1.html"\nprint "Publication(s) related to the potential:"\nprint "https://doi.org/10.1016/j.jnucmat.2015.09.030"\nprint "Parameter file(s) can be downloaded at:"\nprint "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/Al.meam"\nprint "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/library-Al.meam"\npair_style meam\npair_coeff * * library-Al.meam Al Al.meam Al\nmass 1 26.9815\n\n', 'timestep': 0.001, 'temperature': 100, 'pressure': 0.0}
Fill in template
[17]:
# Generate script from template and lammps_variable
script = am.tools.filltemplate(template, lammps_variable, '<', '>')
# Save script to 'nvt.in'
with open('nvt.in', 'w') as f:
f.write(script)
# Show contents of 'nvt.in'
with open('nvt.in') as f:
print(f.read())
units metal
atom_style atomic
boundary p p p
read_data atom.dat
print "Potential 2015--Pascuet-M-I--Al--LAMMPS--ipr1 listed in the NIST Interatomic Potentials Repository:"
print "https://www.ctcms.nist.gov/potentials/entry/2015--Pascuet-M-I-Fernandez-J-R--Al/2015--Pascuet-M-I--Al--LAMMPS--ipr1.html"
print "Publication(s) related to the potential:"
print "https://doi.org/10.1016/j.jnucmat.2015.09.030"
print "Parameter file(s) can be downloaded at:"
print "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/Al.meam"
print "https://www.ctcms.nist.gov/potentials/Download/2015--Pascuet-M-I-Fernandez-J-R--Al/1/library-Al.meam"
pair_style meam
pair_coeff * * library-Al.meam Al Al.meam Al
mass 1 26.9815
# Define temperature and dependent variables
variable T equal 100
variable twoT equal 2*$T
# Define equilibrium pressure
variable P equal 0.0
# Define timestep and dependent variables
variable deltat equal 0.001
variable Trelax equal 100*${deltat}
variable Prelax equal 1000*${deltat}
# Initialize atomic velocities with twoT
velocity all create ${twoT} 124352
# Define thermo steps and properties
thermo 100
thermo_style custom step temp press lx ly lz
# Define dump
dump mydump all atom 100000 *.dump
# Specify timestep
timestep ${deltat}
# Apply npt conditions
fix 1 all npt temp $T $T ${Trelax} iso $P $P ${Prelax}
# Run simulation
run 100000
6. Run LAMMPS
The LAMMPS simulation can be ran from within Python using the run() function.
Specify your lammps executable
[18]:
lammps_exe = '/home/lmh1/LAMMPS/2022-06-23/src/lmp_mpi'
Run the simulation
[19]:
output = lmp.run(lammps_exe, 'nvt.in')
The resulting simulation data is returned as a Log object, which containes the thermo data from the log.lammps files.
7. Analyzing Thermo Results
Data for each simulation run/minimization is stored in the returned Log object. Each Simulation has a thermo property that is a pandas.DataFrame of the LAMMPS thermo data.
Updated version 1.3.7: Each simulation is now represented using a Simulation class rather than a dictionary.
Show thermo data column names for the first (and only) simulation run.
[20]:
print(list(output.simulations[0].thermo.keys()))
['Step', 'Temp', 'Press', 'Lx', 'Ly', 'Lz']
For backwards compatibility, the thermo can also be accessed as if the simulation was still a dictionary.
[21]:
print(list(output.simulations[0]['thermo'].keys()))
['Step', 'Temp', 'Press', 'Lx', 'Ly', 'Lz']
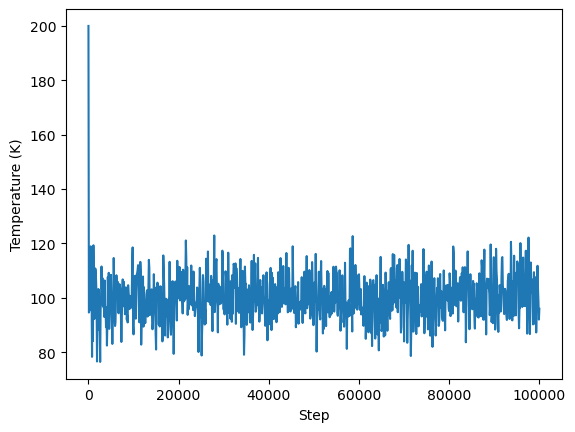
Plot temperature vs. run step
[22]:
# Pull out data
steps = output.simulations[0].thermo.Step
temps = output.simulations[0].thermo.Temp
# Plot
plt.plot(steps, temps)
plt.xlabel('Step')
plt.ylabel('Temperature (K)')
plt.show()
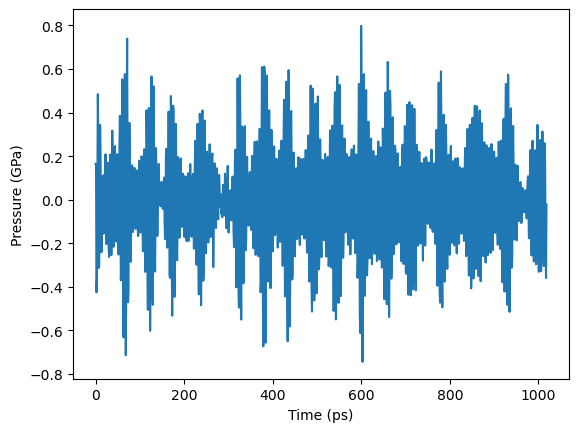
Plot pressure vs. run step (note unit conversions)
[23]:
# Convert steps to time in atomman working units
steps = output.simulations[0].thermo.Step
time = uc.set_in_units(timestep, lammps_unit['time']) * steps
# Convert press to atomman working units
press = uc.set_in_units(output.simulations[0].thermo.Press, lammps_unit['pressure'])
# Plot in ps and GPa
plt.plot(uc.get_in_units(steps, 'ps'), uc.get_in_units(press, 'GPa'))
plt.xlabel('Time (ps)')
plt.ylabel('Pressure (GPa)')
plt.show()
8 Reading in dump files
Finally, any dump files generated by the LAMMPS simulation can be loaded into atomman as Systems.
[24]:
system = am.load('atom_dump', '100000.dump', symbols='Al')
print(system.box)
system.atoms_df()
avect = [12.184, 0.000, 0.000]
bvect = [ 0.000, 12.184, 0.000]
cvect = [ 0.000, 0.000, 12.184]
origin = [-0.017, -0.017, -0.017]
[24]:
| atype | pos[0] | pos[1] | pos[2] | atom_id | |
|---|---|---|---|---|---|
| 0 | 1 | -0.065207 | 0.005483 | -0.067225 | 1 |
| 1 | 1 | 0.015215 | 1.971269 | 2.016629 | 2 |
| 2 | 1 | 1.960085 | -0.049912 | 1.947913 | 3 |
| 3 | 1 | 2.036744 | 2.018590 | 0.026100 | 4 |
| 4 | 1 | 4.021746 | -0.062975 | 0.013582 | 5 |
| ... | ... | ... | ... | ... | ... |
| 103 | 1 | 6.007576 | 10.140925 | 8.201928 | 104 |
| 104 | 1 | 8.188295 | 8.184810 | 8.220727 | 105 |
| 105 | 1 | 8.018944 | 10.181569 | 10.211785 | 106 |
| 106 | 1 | 10.180948 | 8.081238 | 10.182349 | 107 |
| 107 | 1 | 10.151513 | 10.160260 | 8.191060 | 108 |
108 rows × 5 columns
[25]:
am.plot.py3Dmol.view_3d(system)
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
File Cleanup
[27]:
for fname in Path(pot_id).glob('*'):
fname.unlink()
Path('Al.meam').unlink()
Path('library-Al.meam').unlink()
Path('atom.dat').unlink()
Path('nvt.in').unlink()
Path('log.lammps').unlink()
for fname in Path('.').glob('*.dump'):
fname.unlink()